

Characterization and genotype-phenotype correlation of patients with Fanconi anemia in a multi-ethnic population
- PMID: 31558676
- PMCID: PMC7327661
- DOI: 10.3324/haematol.2019.222877
Characterization and genotype-phenotype correlation of patients with Fanconi anemia in a multi-ethnic population
Abstract
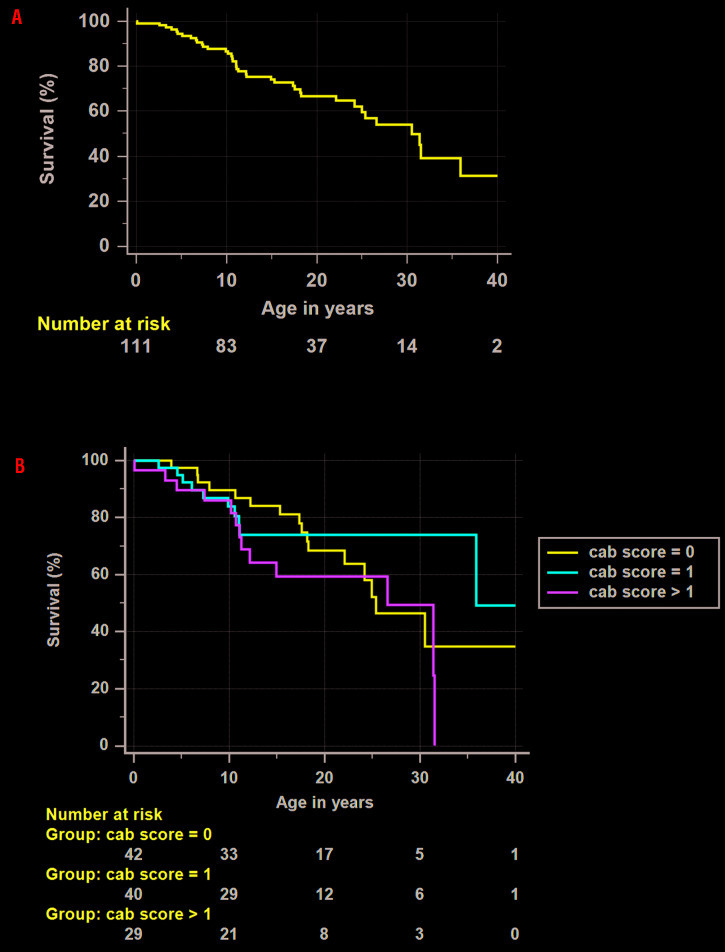
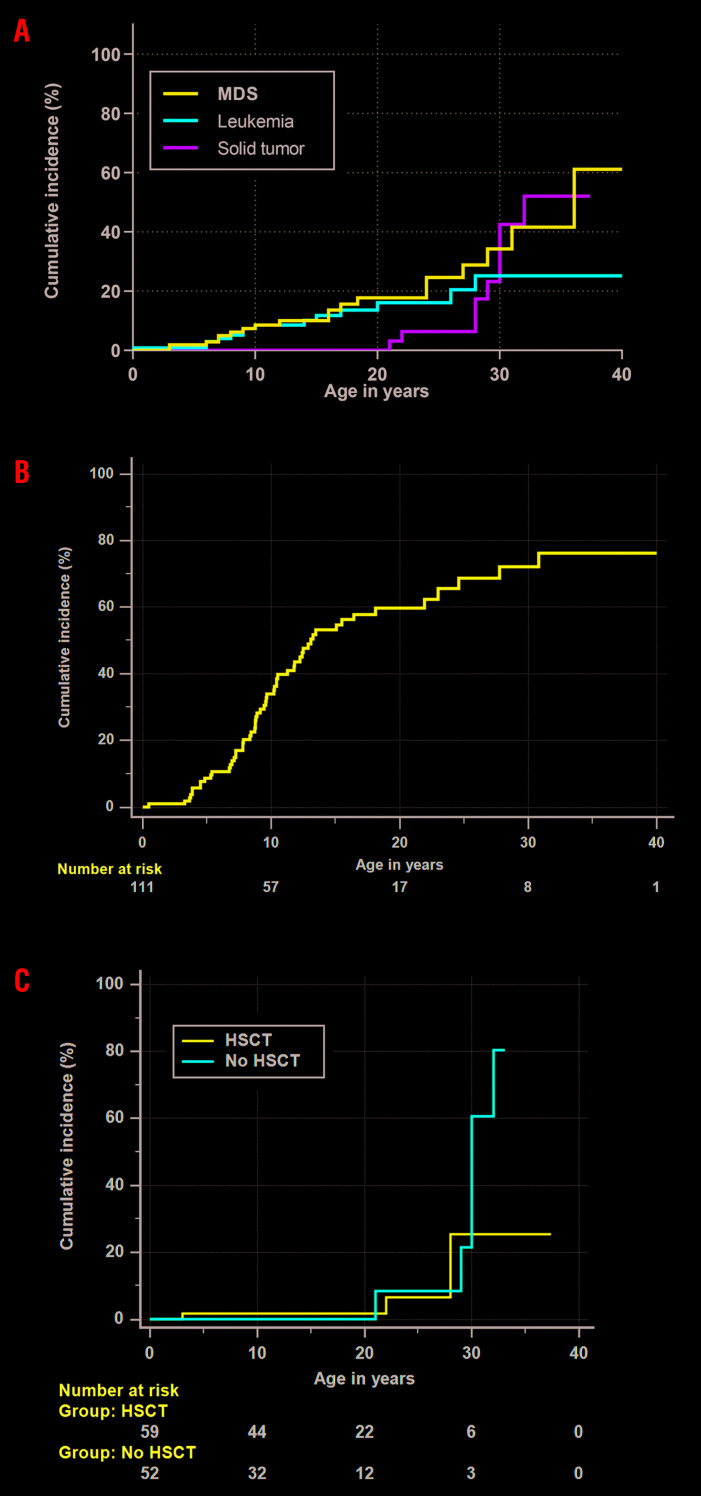
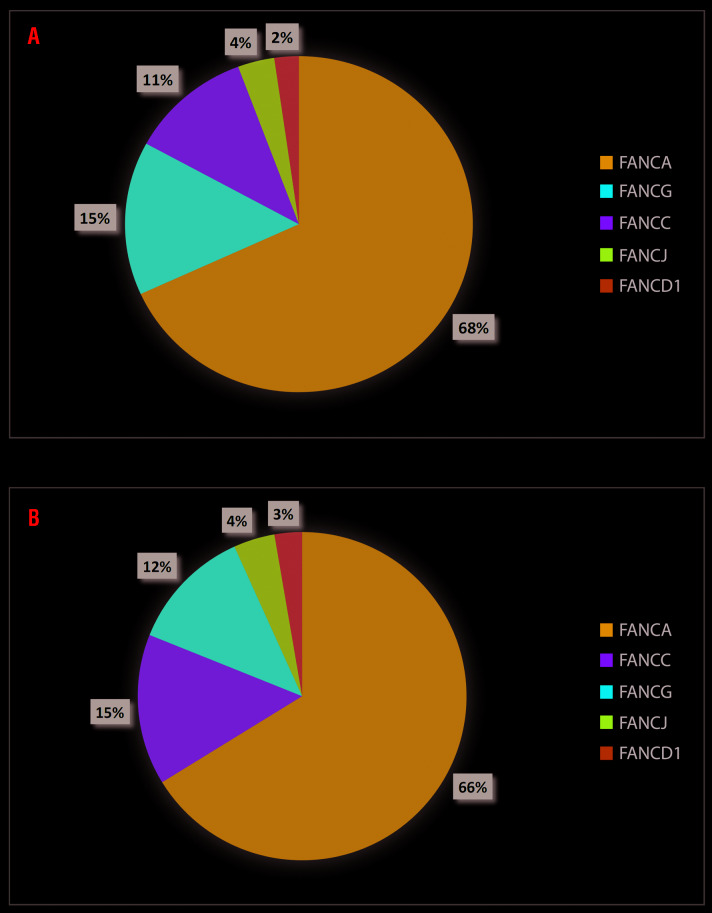
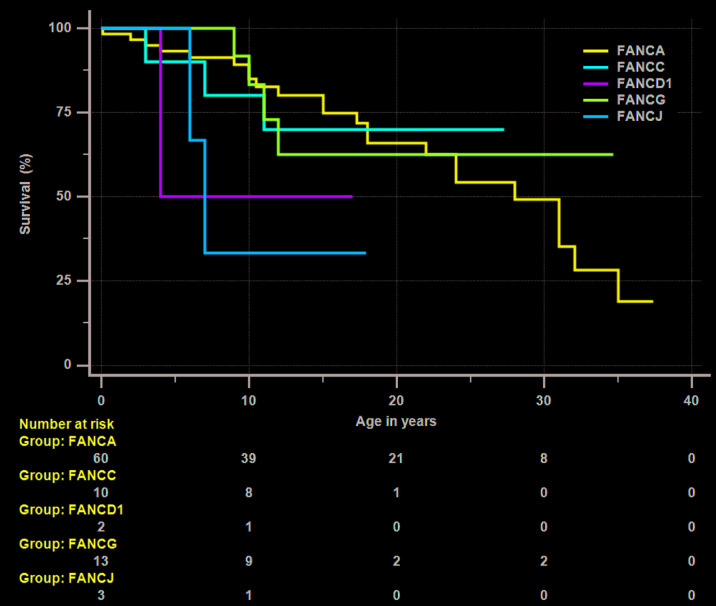
Fanconi anemia (FA), an inherited bone marrow failure (BMF) syndrome, caused by mutations in DNA repair genes, is characterized by congenital anomalies, aplastic anemia, high risk of malignancies and extreme sensitivity to alkylating agents. We aimed to study the clinical presentation, molecular diagnosis and genotype-phenotype correlation among patients with FA from the Israeli inherited BMF registry. Overall, 111 patients of Arab (57%) and Jewish (43%) descent were followed for a median of 15 years (range: 0.1-49); 63% were offspring of consanguineous parents. One-hundred patients (90%) had at least one congenital anomaly; over 80% of the patients developed bone marrow failure; 53% underwent hematopoietic stem-cell transplantation; 33% of the patients developed cancer; no significant association was found between hematopoietic stem-cell transplant and solid tumor development. Nearly 95% of the patients tested had confirmed mutations in the Fanconi genes FANCA (67%), FANCC (13%), FANCG (14%), FANCJ (3%) and FANCD1 (2%), including twenty novel mutations. Patients with FANCA mutations developed cancer at a significantly older age compared to patients with mutations in other Fanconi genes (mean 18.5 and 5.2 years, respectively, P=0.001); however, the overall survival did not depend on the causative gene. We hereby describe a large national cohort of patients with FA, the vast majority genetically diagnosed. Our results suggest an older age for cancer development in patients with FANCA mutations and no increased incidence of solid tumors following hematopoietic stem-cell transplant. Further studies are needed to guide individual treatment and follow-up programs.
Copyright© 2020 Ferrata Storti Foundation.
Figures
References
-
- Callen E, Casado JA, Tischkowitz MD, et al. A common founder mutation in FANCA underlies the world's highest prevalence of Fanconi anemia in Gypsy families from Spain. Blood. 2005;105(5):1946-1949. - PubMed
-
- Morgan NV, Essop F, Demuth I, et al. A common Fanconi anemia mutation in black populations of sub-Saharan Africa. Blood. 2005;105(9):3542-3544. - PubMed
-
- Rosendorff J, Bernstein R, Macdougall L, Jenkins T. Fanconi anemia: another disease of unusually high prevalence in the Afrikaans population of South Africa. Am J Med Genet. 1987;27(4):793-797. - PubMed
-
- Whitney MA, Jakobs P, Kaback M, Moses RE, Grompe M. The Ashkenazi Jewish Fanconi anemia mutation: incidence among patients and carrier frequency in the at-risk population. Hum Mutat. 1994;3(4):339-341. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
