

Exploring the Effectiveness of Binding Free Energy Calculations
- PMID: 31560539
- PMCID: PMC7032235
- DOI: 10.1021/acs.jpcb.9b07593
Exploring the Effectiveness of Binding Free Energy Calculations
Abstract
Increasing the accuracy of the evaluation of ligand-binding energies is one of the most important tasks of current computational biology. Here we explore the accuracy of free energy perturbation (FEP) approaches by comparing the performance of a "regular" FEP method to the one using replica exchange to enhance the sampling on a well-defined benchmark. The examination was limited to the so-called alchemical perturbations which are restricted to a fragment of the drug, and therefore, the calculation is a relative one rather than the absolute binding energy of the drug. Overall, our calculations reach the 1 kcal/mol accuracy limit. It is also shown that the accurate prediction of the position of water molecules around the binding pocket is important for FEP calculations. Interestingly, the replica exchange method does not significantly improve the accuracy of binding energies, suggesting that we reach the limit where the force field quality is a critical factor for accurate calculations.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
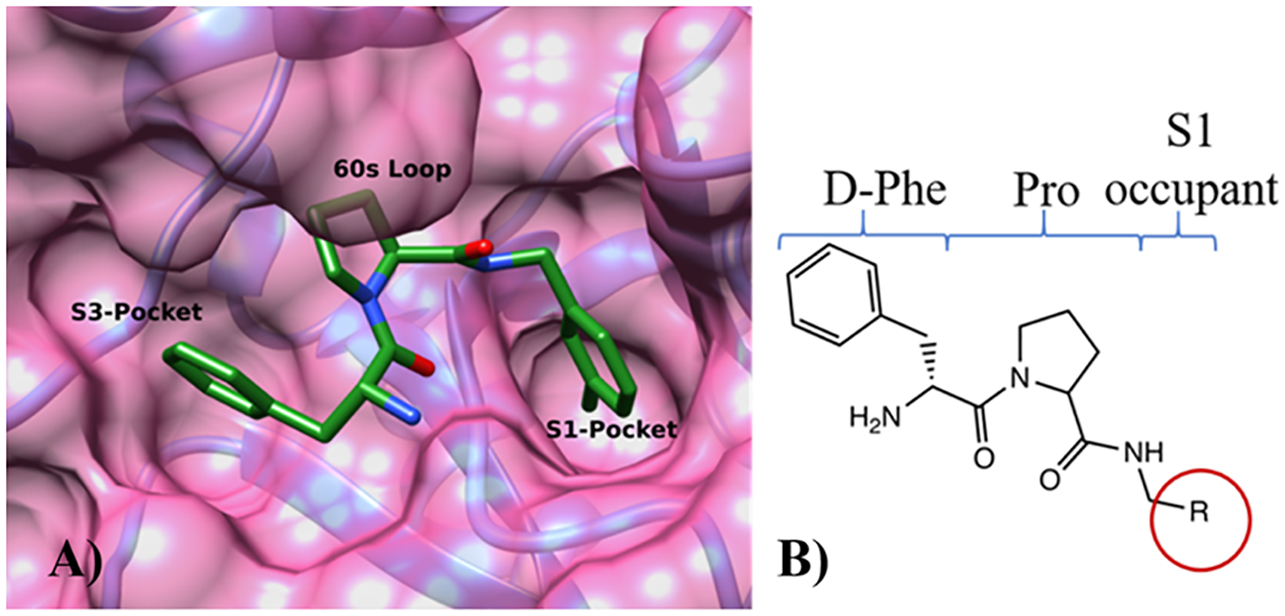




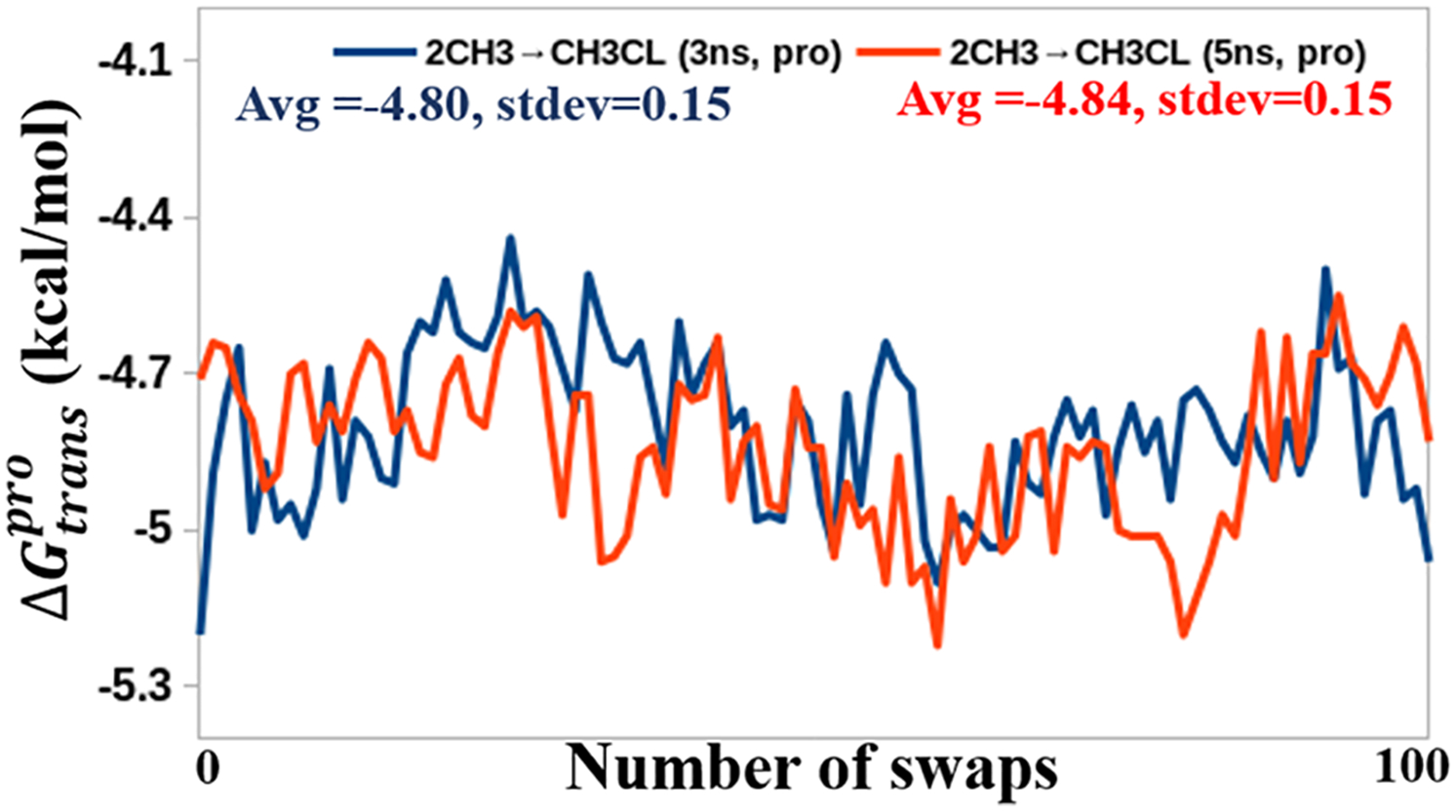


References
-
- Wang L; Wu Y; Deng Y; Kim B; Pierce L; Krilov G; Lupyan D; Robinson S; Dahlgren MK; Greenwood J; et al. Accurate and reliable prediction of relative ligand binding potency in prospective drug discovery by way of a modern free-energy calculation protocol and force field. J. Am. Chem. Soc 2015, 137, 2695–2703. - PubMed
-
- Cournia Z; Allen B; Sherman W Relative binding free energy calculations in drug discovery: recent advances and practical considerations. J. Chem. Inf. Model 2017, 57, 2911–2937. - PubMed
-
- Torrie GM; Valleau JP Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys 1977, 23, 187–199.
-
- Sugita Y; Okamoto Y Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett 1999, 314, 141–151.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
