

Autosomal-Recessive Mutations in MESD Cause Osteogenesis Imperfecta
- PMID: 31564437
- PMCID: PMC6817720
- DOI: 10.1016/j.ajhg.2019.08.008
Autosomal-Recessive Mutations in MESD Cause Osteogenesis Imperfecta
Abstract
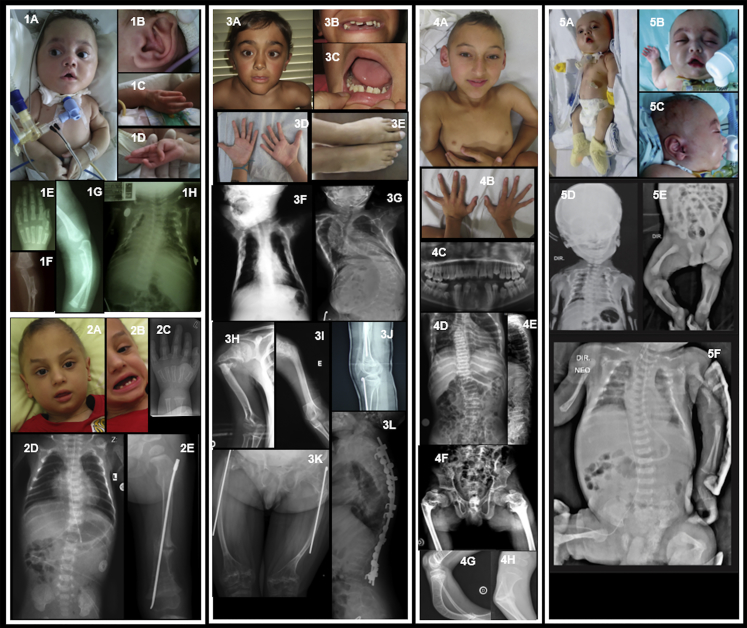
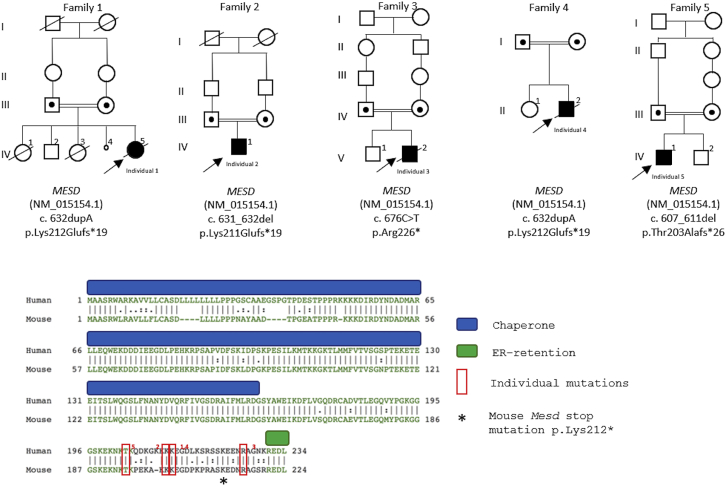
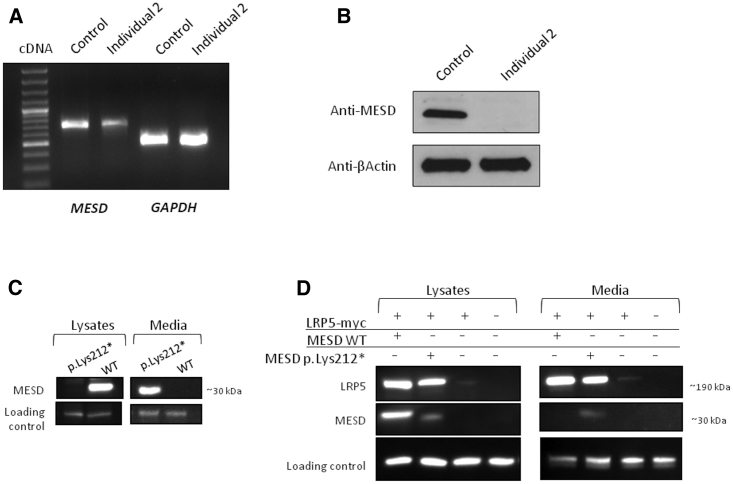
Osteogenesis imperfecta (OI) comprises a genetically heterogeneous group of skeletal fragility diseases. Here, we report on five independent families with a progressively deforming type of OI, in whom we identified four homozygous truncation or frameshift mutations in MESD. Affected individuals had recurrent fractures and at least one had oligodontia. MESD encodes an endoplasmic reticulum (ER) chaperone protein for the canonical Wingless-related integration site (WNT) signaling receptors LRP5 and LRP6. Because complete absence of MESD causes embryonic lethality in mice, we hypothesized that the OI-associated mutations are hypomorphic alleles since these mutations occur downstream of the chaperone activity domain but upstream of ER-retention domain. This would be consistent with the clinical phenotypes of skeletal fragility and oligodontia in persons deficient for LRP5 and LRP6, respectively. When we expressed wild-type (WT) and mutant MESD in HEK293T cells, we detected WT MESD in cell lysate but not in conditioned medium, whereas the converse was true for mutant MESD. We observed that both WT and mutant MESD retained the ability to chaperone LRP5. Thus, OI-associated MESD mutations produce hypomorphic alleles whose failure to remain within the ER significantly reduces but does not completely eliminate LRP5 and LRP6 trafficking. Since these individuals have no eye abnormalities (which occur in individuals completely lacking LRP5) and have neither limb nor brain patterning defects (both of which occur in mice completely lacking LRP6), we infer that bone mass accrual and dental patterning are more sensitive to reduced canonical WNT signaling than are other developmental processes. Biologic agents that can increase LRP5 and LRP6-mediated WNT signaling could benefit individuals with MESD-associated OI.
Keywords: MESD; WNT signaling; osteogenesis imperfecta.
Copyright © 2019 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Marini J.C., Forlino A., Bächinger H.P., Bishop N.J., Byers P.H., Paepe A., Fassier F., Fratzl-Zelman N., Kozloff K.M., Krakow D. Osteogenesis imperfecta. Nat. Rev. Dis. Primers. 2017;3:17052. - PubMed
-
- Holdener B.C., Faust C., Rosenthal N.S., Magnuson T. msd is required for mesoderm induction in mice. Development. 1994;120:1335–1346. - PubMed
-
- Wines M.E., Shi Y., Lindor M., Holdener B.C. Physical localization of the mesoderm development (mesd) functional region. Genomics. 2000;68:322–329. - PubMed
-
- Hsieh J.C., Lee L., Zhang L., Wefer S., Brown K., DeRossi C., Wines M.E., Rosenquist T., Holdener B.C. Mesd encodes an LRP5/6 chaperone essential for specification of mouse embryonic polarity. Cell. 2003;112:355–367. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
