

An Evolutionarily Conserved Function of Polycomb Silences the MHC Class I Antigen Presentation Pathway and Enables Immune Evasion in Cancer
- PMID: 31564637
- PMCID: PMC6876280
- DOI: 10.1016/j.ccell.2019.08.008
An Evolutionarily Conserved Function of Polycomb Silences the MHC Class I Antigen Presentation Pathway and Enables Immune Evasion in Cancer
Abstract
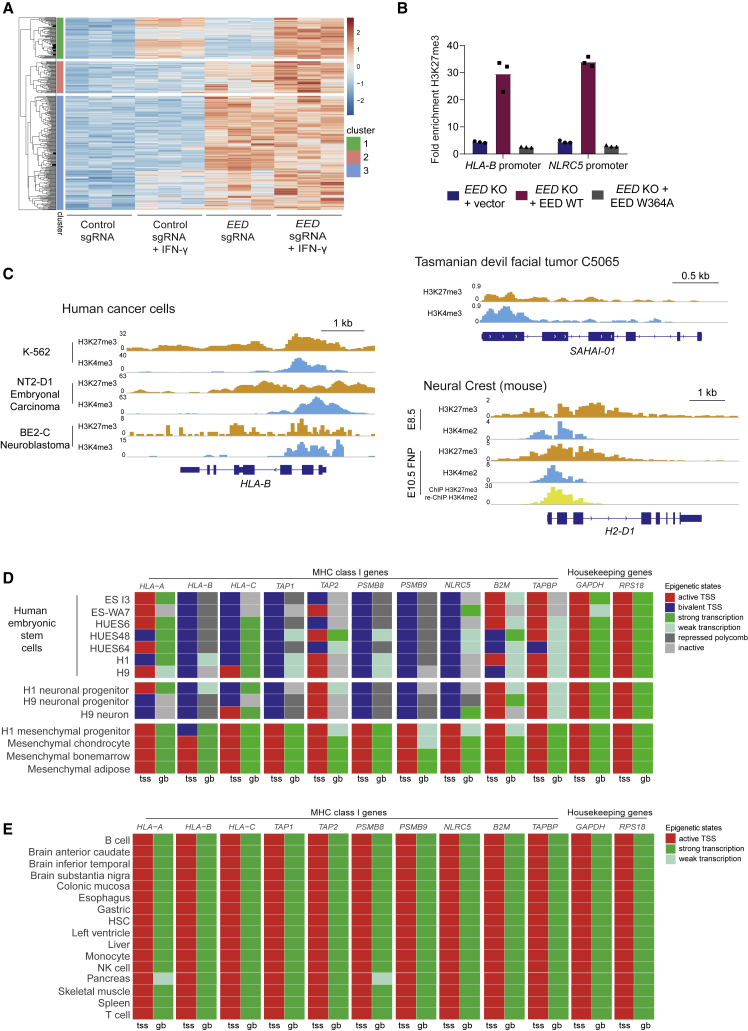
Loss of MHC class I (MHC-I) antigen presentation in cancer cells can elicit immunotherapy resistance. A genome-wide CRISPR/Cas9 screen identified an evolutionarily conserved function of polycomb repressive complex 2 (PRC2) that mediates coordinated transcriptional silencing of the MHC-I antigen processing pathway (MHC-I APP), promoting evasion of T cell-mediated immunity. MHC-I APP gene promoters in MHC-I low cancers harbor bivalent activating H3K4me3 and repressive H3K27me3 histone modifications, silencing basal MHC-I expression and restricting cytokine-induced upregulation. Bivalent chromatin at MHC-I APP genes is a normal developmental process active in embryonic stem cells and maintained during neural progenitor differentiation. This physiological MHC-I silencing highlights a conserved mechanism by which cancers arising from these primitive tissues exploit PRC2 activity to enable immune evasion.
Keywords: EZH2; MHC class I; antigen presentation; bivalency; cancer; epigenetic repression; histone methyltransferase; immune evasion; immunotherapy; polycomb.
Copyright © 2019 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
M.T.M. and R.K.P. are employees of GlaxoSmithKline. M.A.D. has been a member of advisory boards for CTX CRC, Storm Therapeutics, Celgene, and Cambridge Epigenetix.
Figures








Comment in
-
PRC2 Plays Red Light, Green Light with MHC-I and CD8+ T Cells.Cancer Cell. 2019 Oct 14;36(4):343-345. doi: 10.1016/j.ccell.2019.09.010. Cancer Cell. 2019. PMID: 31614112
-
PRC2-mediated MHC-I silencing drives immune evasion.Nat Rev Cancer. 2019 Dec;19(12):664. doi: 10.1038/s41568-019-0219-4. Nat Rev Cancer. 2019. PMID: 31616077 No abstract available.
References
-
- Aggarwal R., Huang J., Alumkal J.J., Zhang L., Feng F.Y., Thomas G.V., Weinstein A.S., Friedl V., Zhang C., Witte O.N. Clinical and genomic characterization of treatment-emergent small-cell neuroendocrine prostate cancer: a multi-institutional prospective study. J. Clin. Oncol. 2018;36:2492–2503. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
