

Telescope: Characterization of the retrotranscriptome by accurate estimation of transposable element expression
- PMID: 31568525
- PMCID: PMC6786656
- DOI: 10.1371/journal.pcbi.1006453
Telescope: Characterization of the retrotranscriptome by accurate estimation of transposable element expression
Abstract
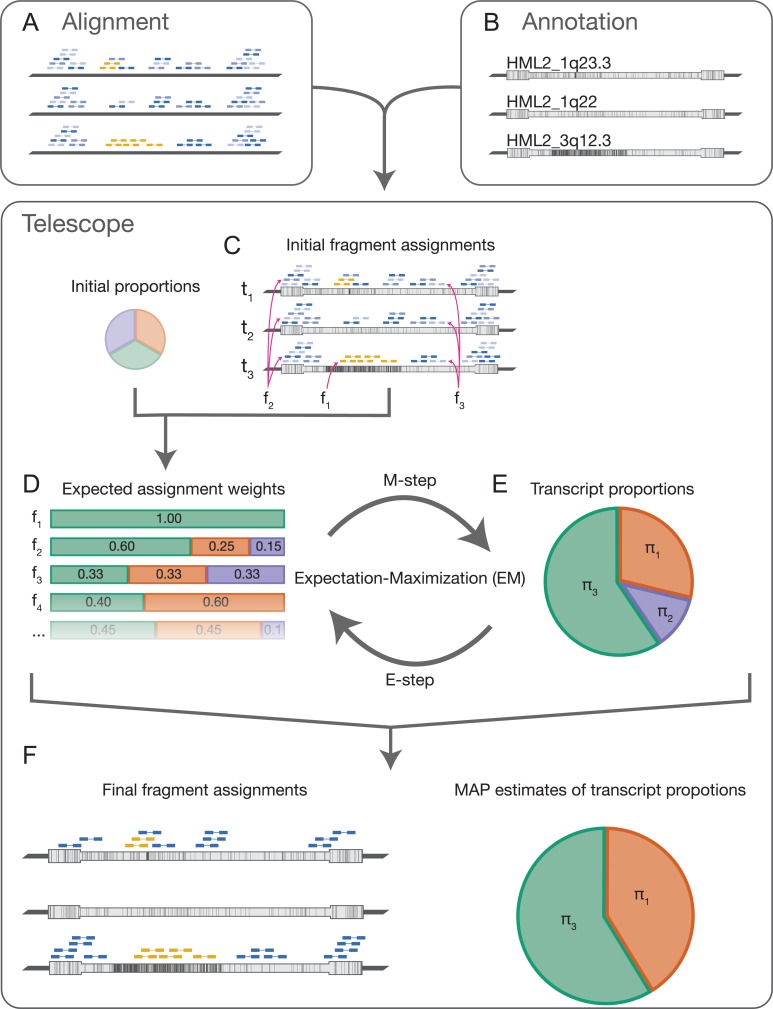
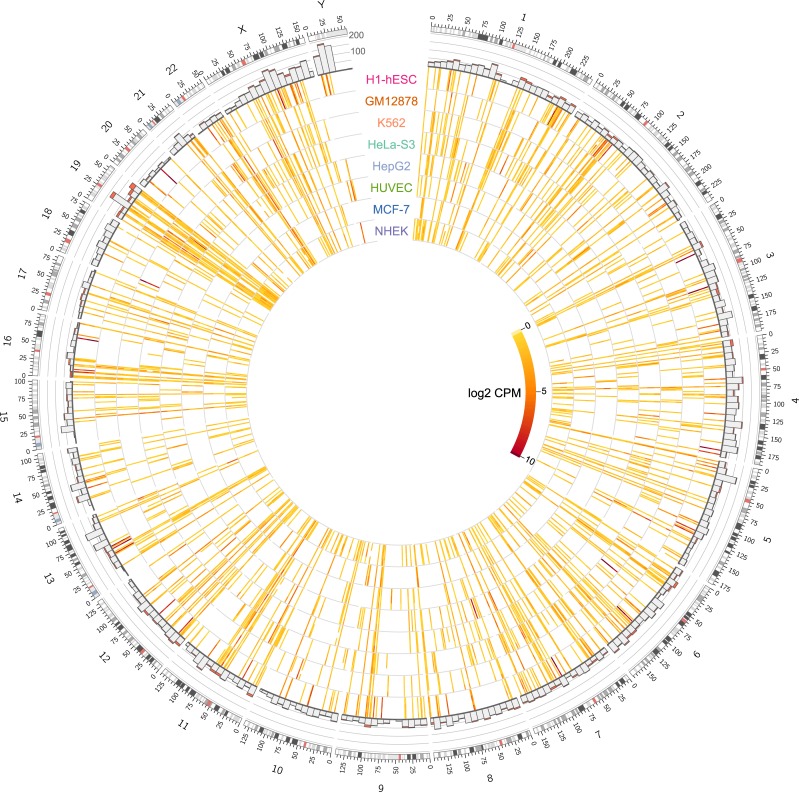
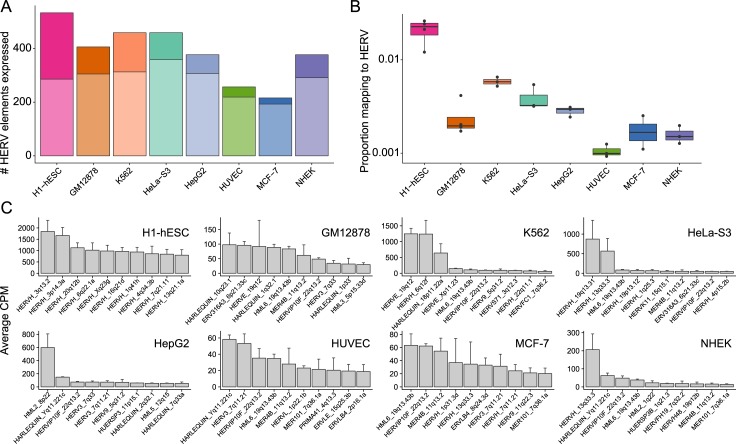
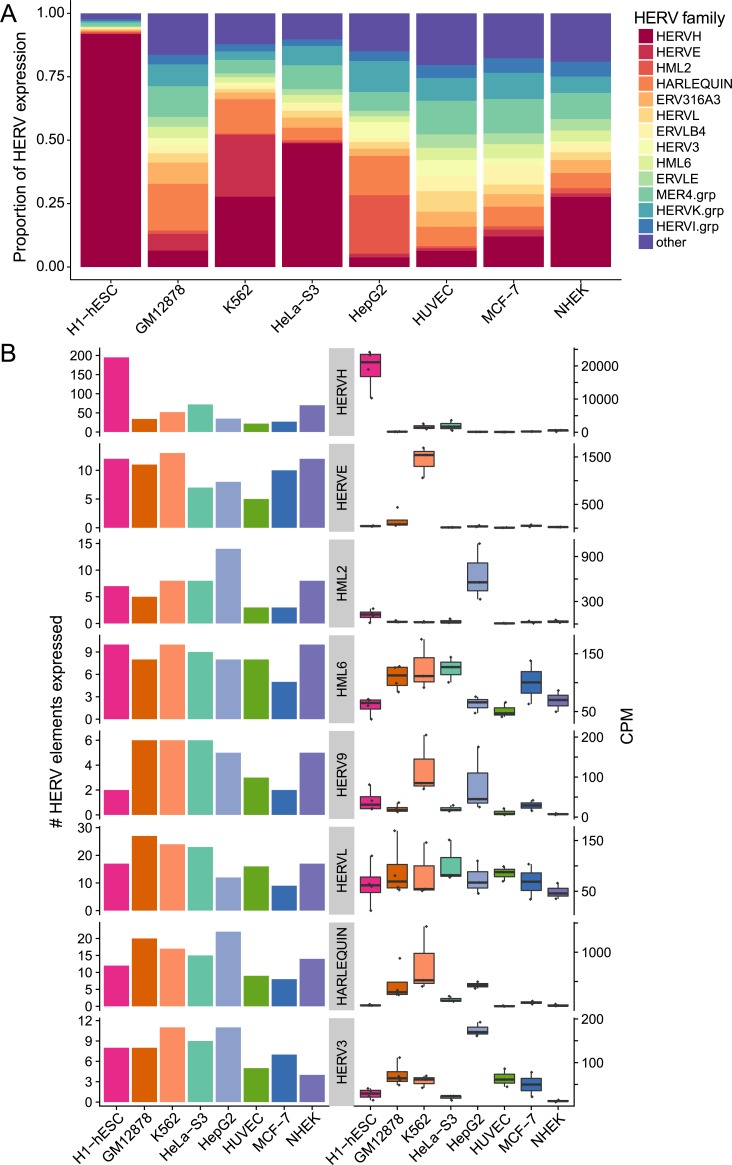
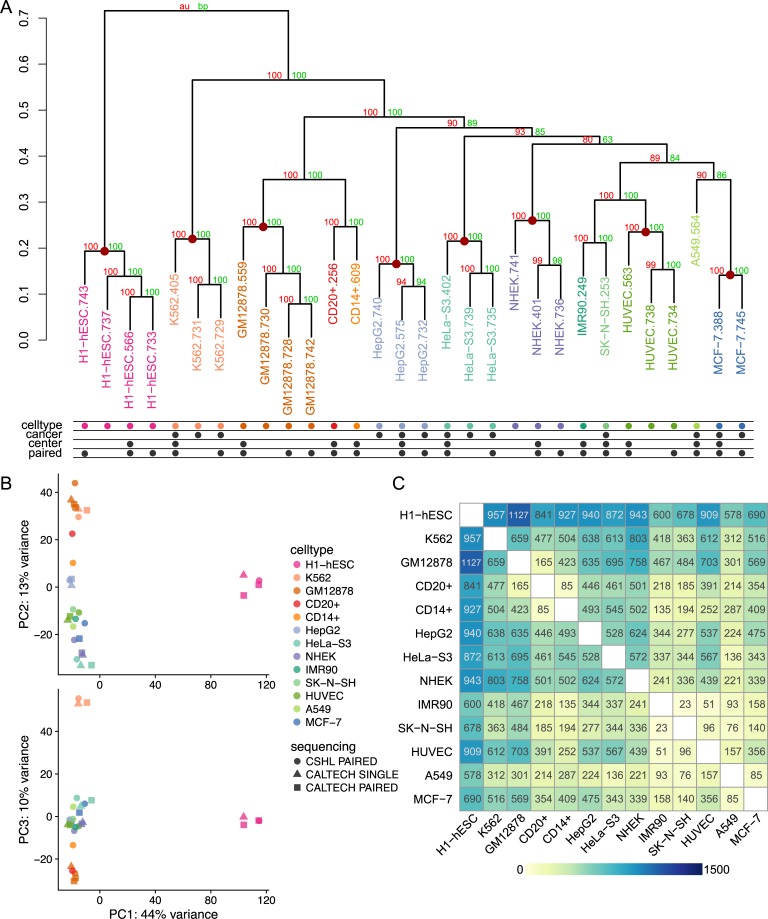
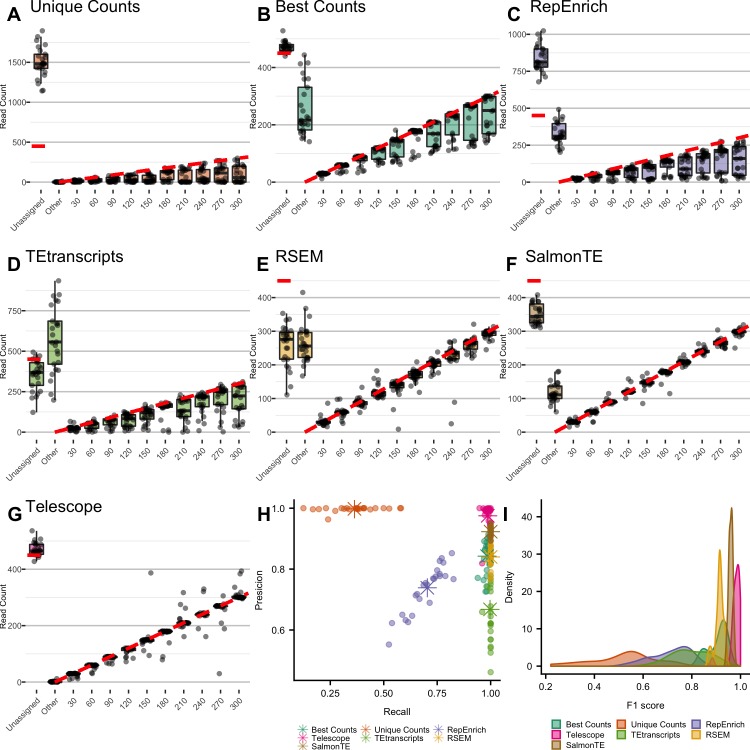
Characterization of Human Endogenous Retrovirus (HERV) expression within the transcriptomic landscape using RNA-seq is complicated by uncertainty in fragment assignment because of sequence similarity. We present Telescope, a computational software tool that provides accurate estimation of transposable element expression (retrotranscriptome) resolved to specific genomic locations. Telescope directly addresses uncertainty in fragment assignment by reassigning ambiguously mapped fragments to the most probable source transcript as determined within a Bayesian statistical model. We demonstrate the utility of our approach through single locus analysis of HERV expression in 13 ENCODE cell types. When examined at this resolution, we find that the magnitude and breadth of the retrotranscriptome can be vastly different among cell types. Furthermore, our approach is robust to differences in sequencing technology and demonstrates that the retrotranscriptome has potential to be used for cell type identification. We compared our tool with other approaches for quantifying transposable element (TE) expression, and found that Telescope has the greatest resolution, as it estimates expression at specific TE insertions rather than at the TE subfamily level. Telescope performs highly accurate quantification of the retrotranscriptomic landscape in RNA-seq experiments, revealing a differential complexity in the transposable element biology of complex systems not previously observed. Telescope is available at https://github.com/mlbendall/telescope.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
SARS-CoV-2 infection mediates differential expression of human endogenous retroviruses and long interspersed nuclear elements.JCI Insight. 2021 Dec 22;6(24):e147170. doi: 10.1172/jci.insight.147170. JCI Insight. 2021. PMID: 34731091 Free PMC article.
-
Human Endogenous Retrovirus Expression Is Associated with Head and Neck Cancer and Differential Survival.Viruses. 2020 Aug 28;12(9):956. doi: 10.3390/v12090956. Viruses. 2020. PMID: 32872377 Free PMC article.
-
SQuIRE reveals locus-specific regulation of interspersed repeat expression.Nucleic Acids Res. 2019 Mar 18;47(5):e27. doi: 10.1093/nar/gky1301. Nucleic Acids Res. 2019. PMID: 30624635 Free PMC article.
-
New insight into transcription of human endogenous retroviral elements.N Biotechnol. 2013 Mar 25;30(3):314-8. doi: 10.1016/j.nbt.2012.11.009. Epub 2012 Nov 29. N Biotechnol. 2013. PMID: 23201072 Review.
-
Utility of next-generation RNA-sequencing in identifying chimeric transcription involving human endogenous retroviruses.APMIS. 2016 Jan-Feb;124(1-2):127-39. doi: 10.1111/apm.12477. APMIS. 2016. PMID: 26818267 Review.
Cited by
-
Locus-Specific Characterization of Human Endogenous Retrovirus Expression in Prostate, Breast, and Colon Cancers.Cancer Res. 2021 Jul 1;81(13):3449-3460. doi: 10.1158/0008-5472.CAN-20-3975. Epub 2021 May 3. Cancer Res. 2021. PMID: 33941616 Free PMC article.
-
Comprehensive Analysis of Large-Scale Transcriptomes from Multiple Cancer Types.Genes (Basel). 2021 Nov 24;12(12):1865. doi: 10.3390/genes12121865. Genes (Basel). 2021. PMID: 34946814 Free PMC article.
-
Human reproduction is regulated by retrotransposons derived from ancient Hominidae-specific viral infections.Nat Commun. 2022 Jan 24;13(1):463. doi: 10.1038/s41467-022-28105-1. Nat Commun. 2022. PMID: 35075135 Free PMC article.
-
Cell-Specific Transposable Element and Gene Expression Analysis Across Systemic Lupus Erythematosus Phenotypes.ACR Open Rheumatol. 2024 Nov;6(11):769-779. doi: 10.1002/acr2.11713. Epub 2024 Aug 14. ACR Open Rheumatol. 2024. PMID: 39143499 Free PMC article.
-
Endogenous retroelements in hematological malignancies: From epigenetic dysregulation to therapeutic targeting.Am J Hematol. 2025 Jan;100(1):116-130. doi: 10.1002/ajh.27501. Epub 2024 Oct 10. Am J Hematol. 2025. PMID: 39387681 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
