

The future of cystic fibrosis care: a global perspective
- PMID: 31570318
- PMCID: PMC8862661
- DOI: 10.1016/S2213-2600(19)30337-6
The future of cystic fibrosis care: a global perspective
Erratum in
-
Correction to Lancet Respir Med 2019; published online Sept 27. https://doi.org/10.1016/S2213-2600(19)30337-6.Lancet Respir Med. 2019 Dec;7(12):e40. doi: 10.1016/S2213-2600(19)30408-4. Epub 2019 Oct 25. Lancet Respir Med. 2019. PMID: 31669225 No abstract available.
Abstract
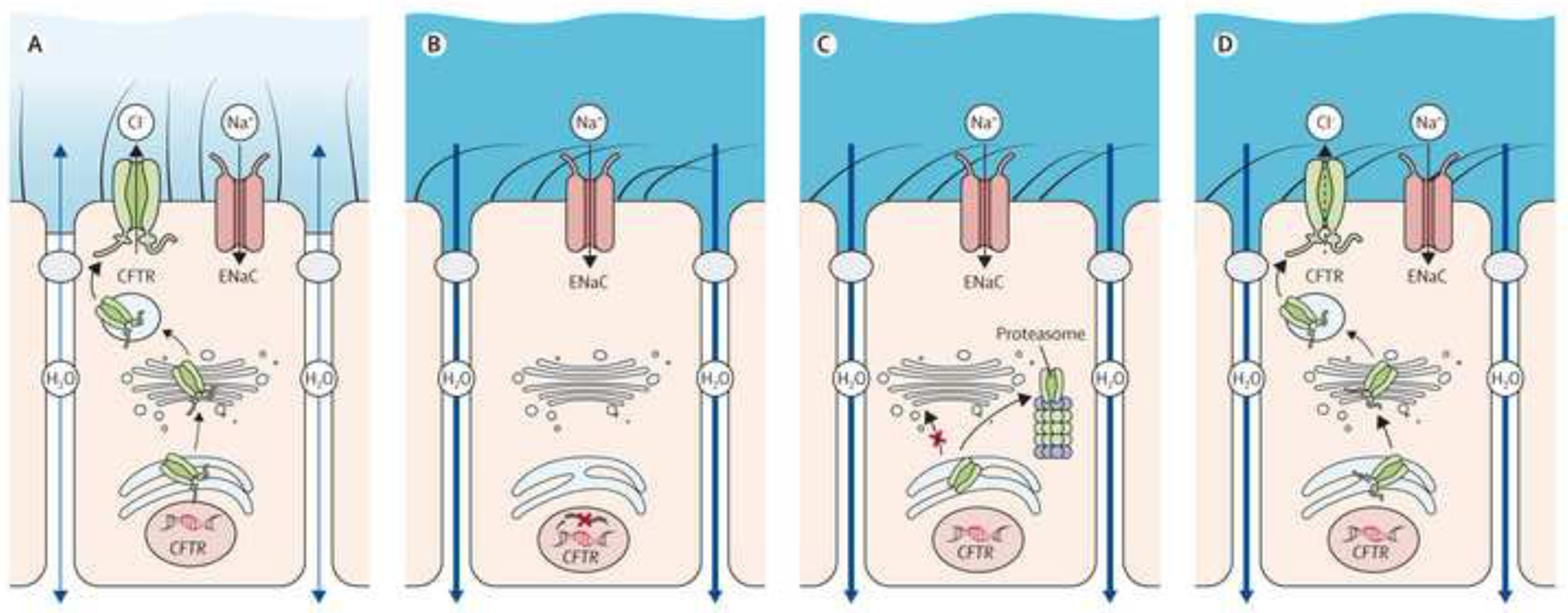
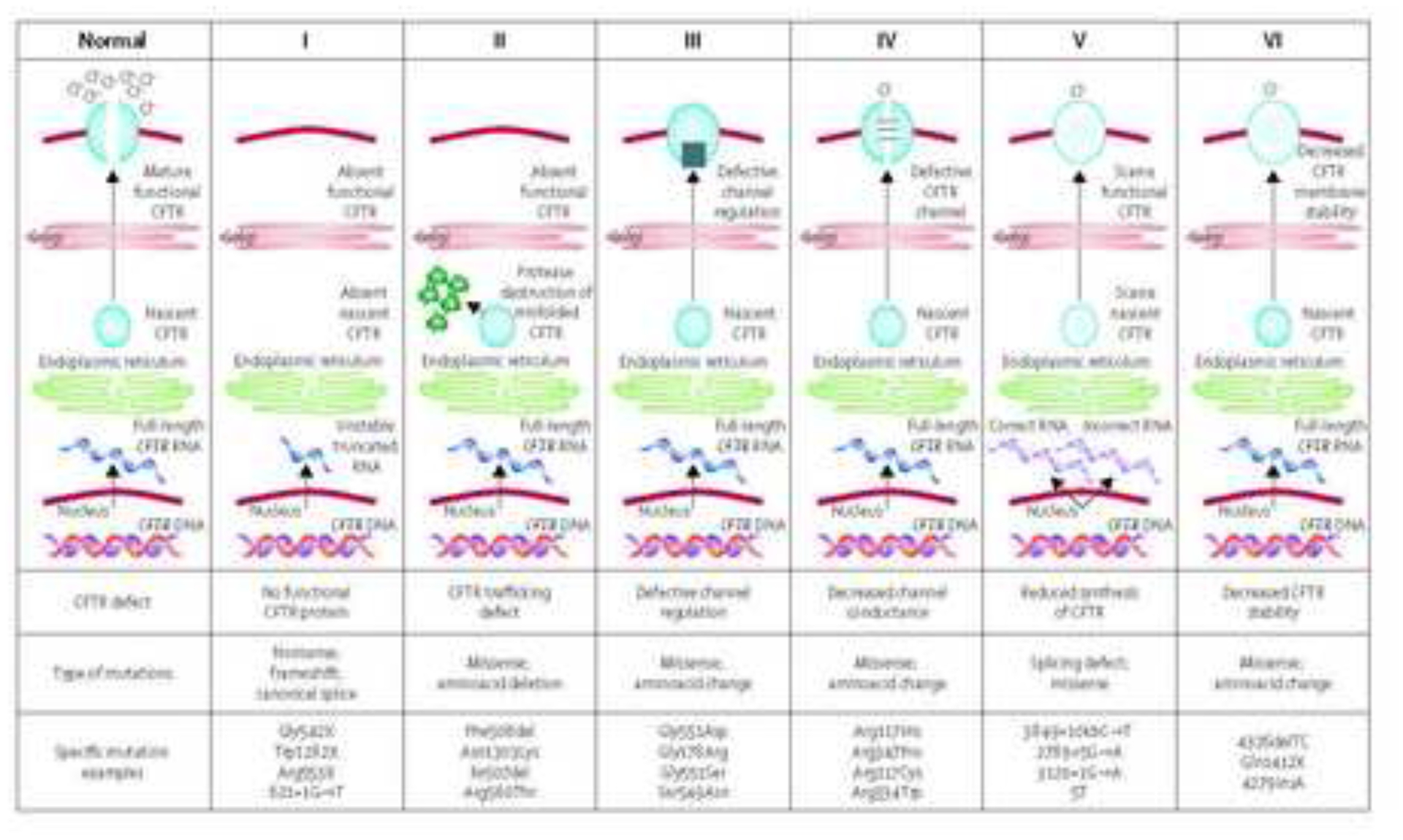
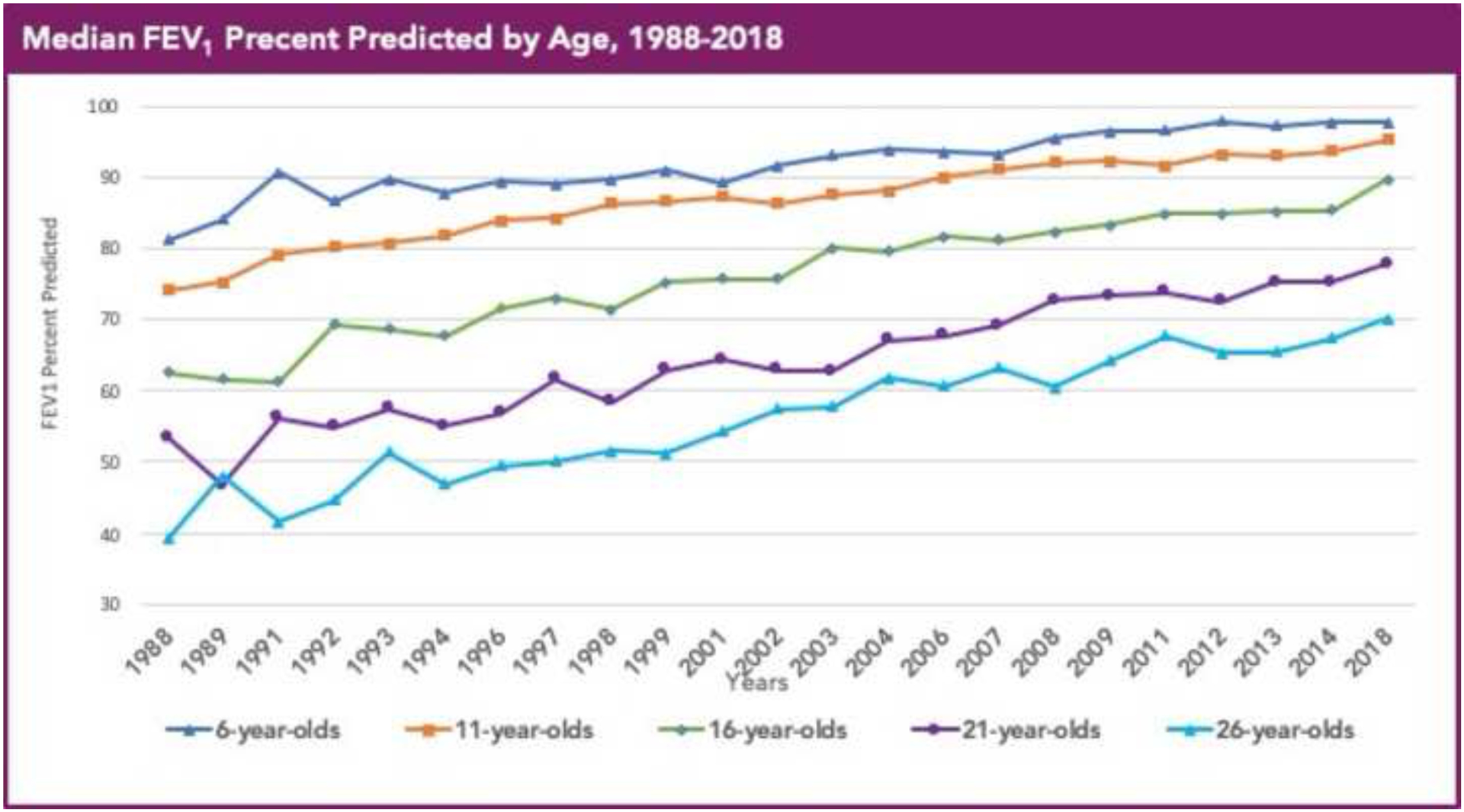
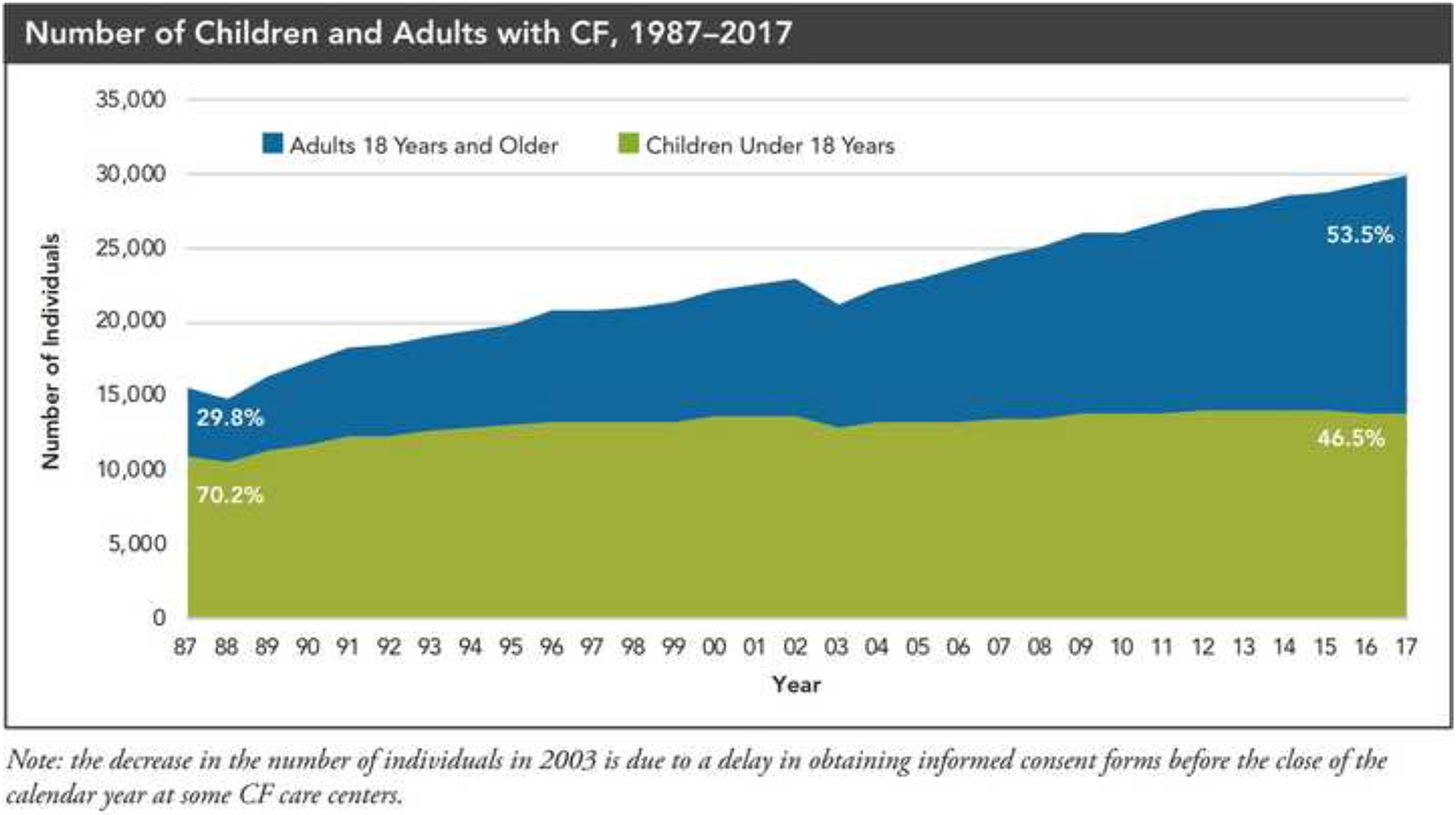
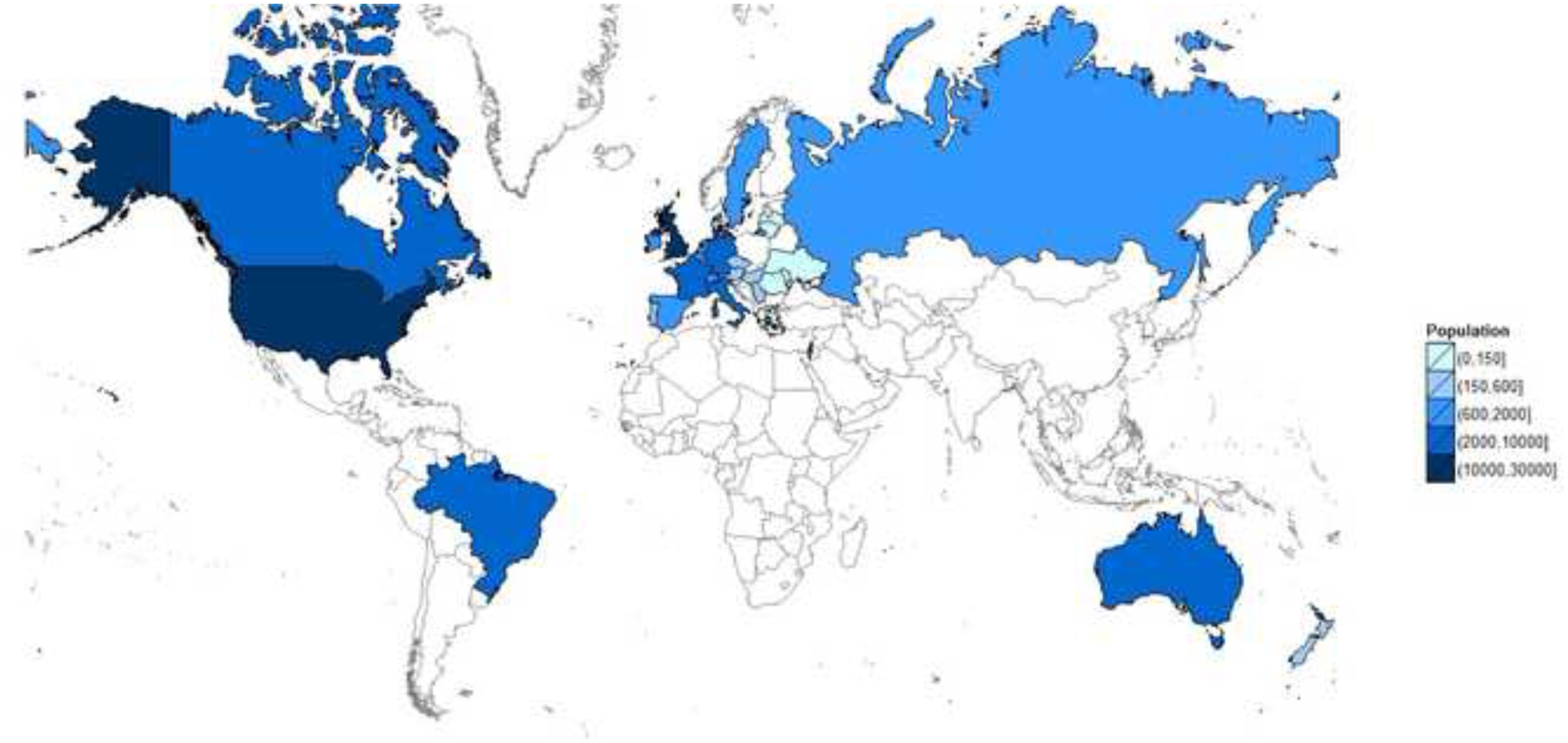
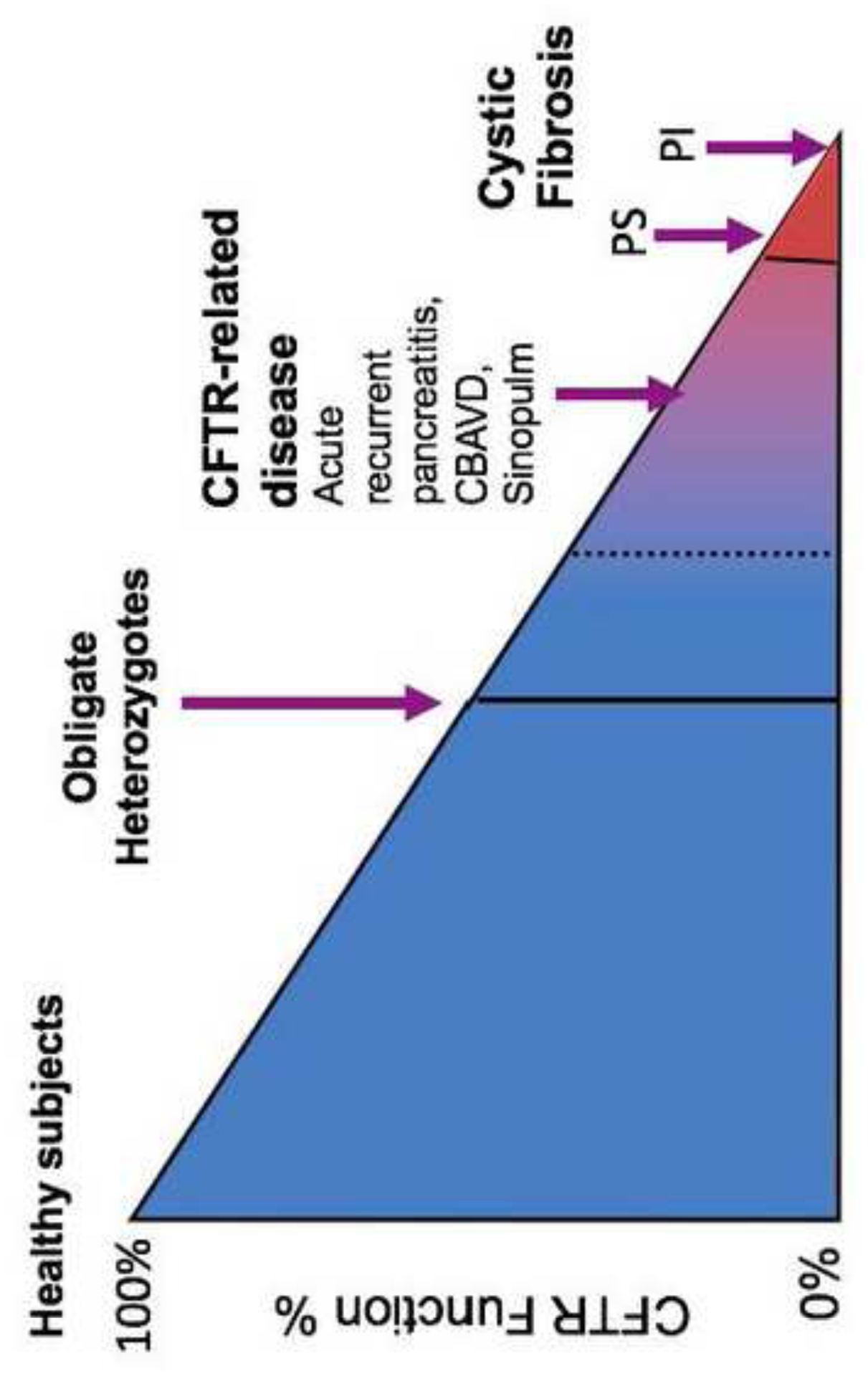
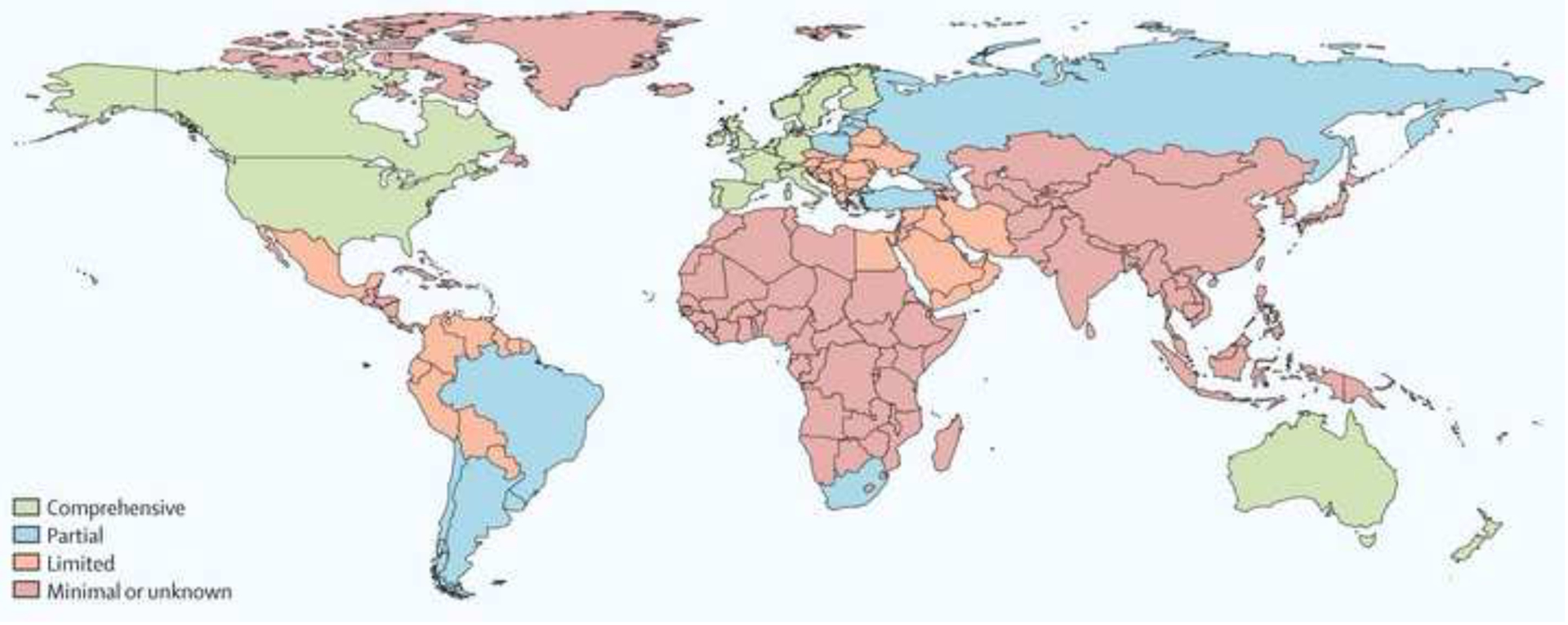
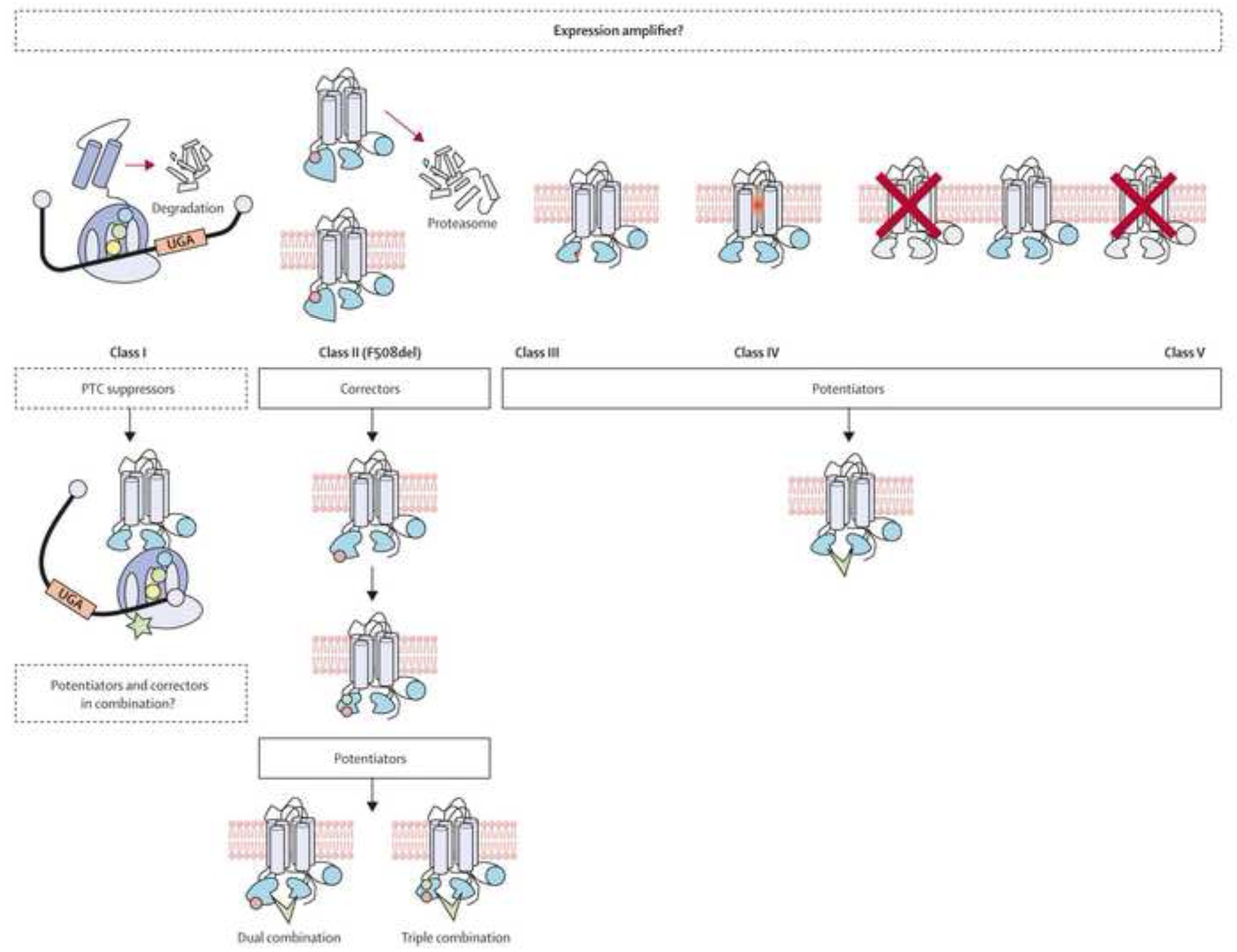
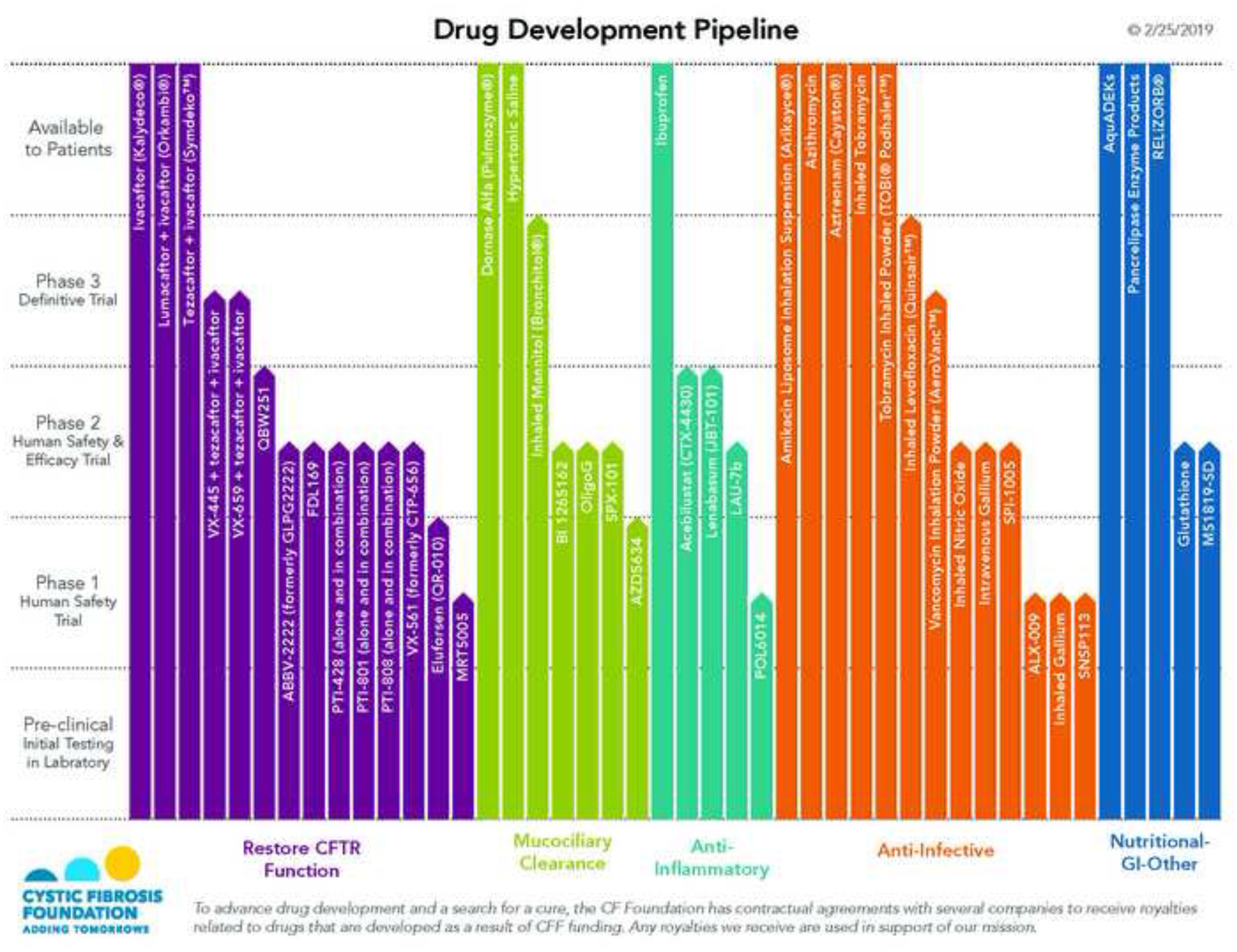
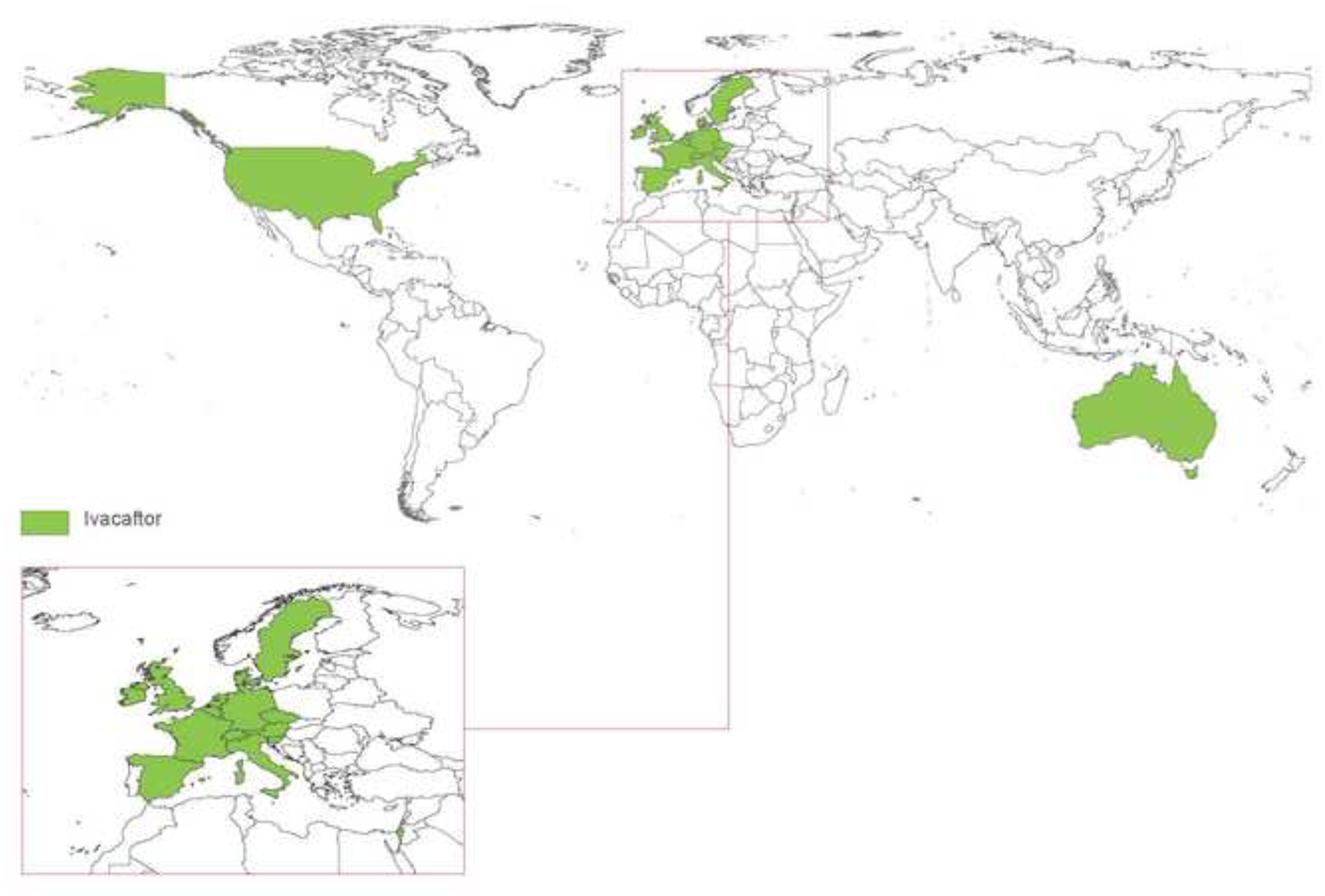
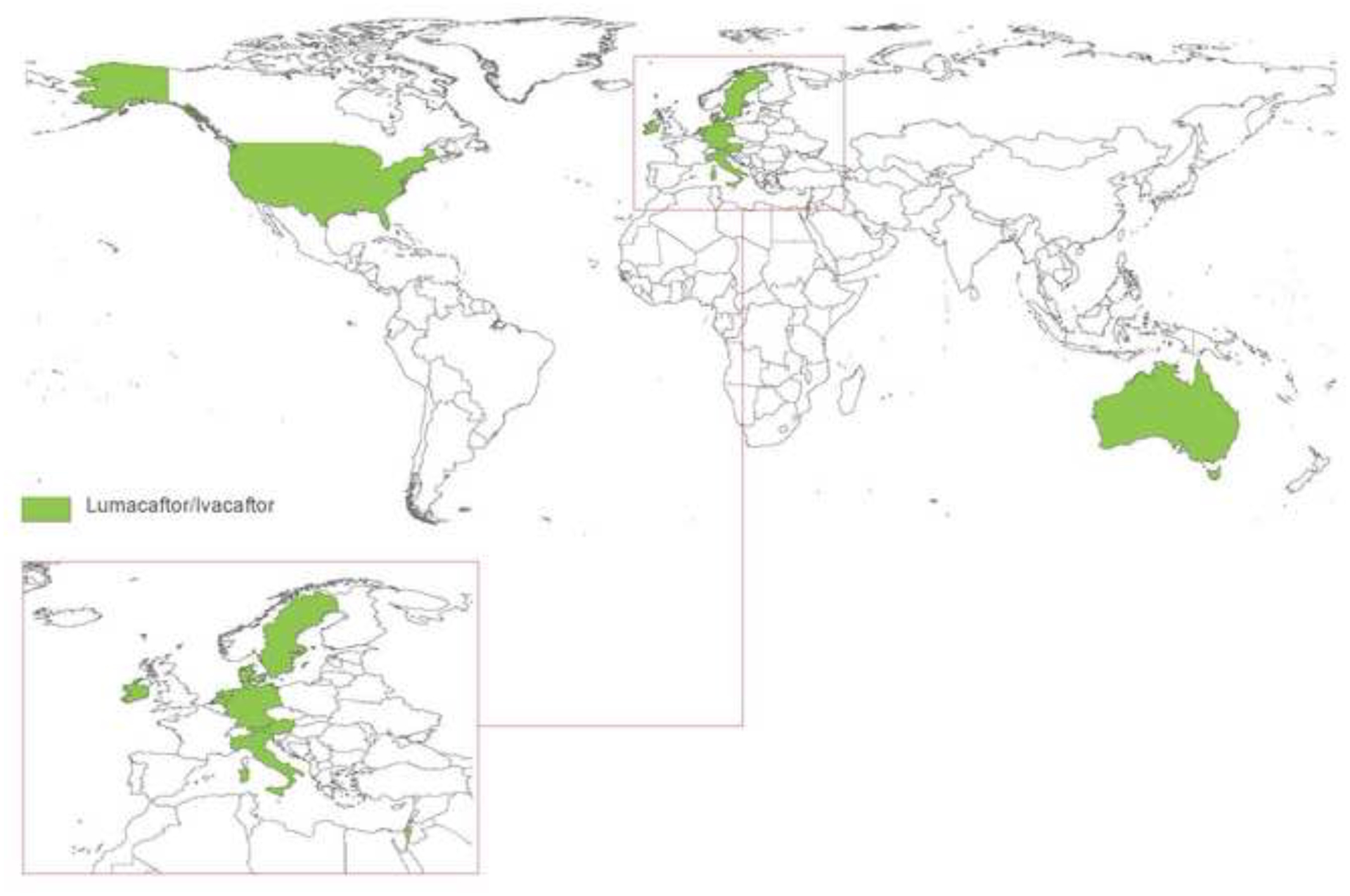
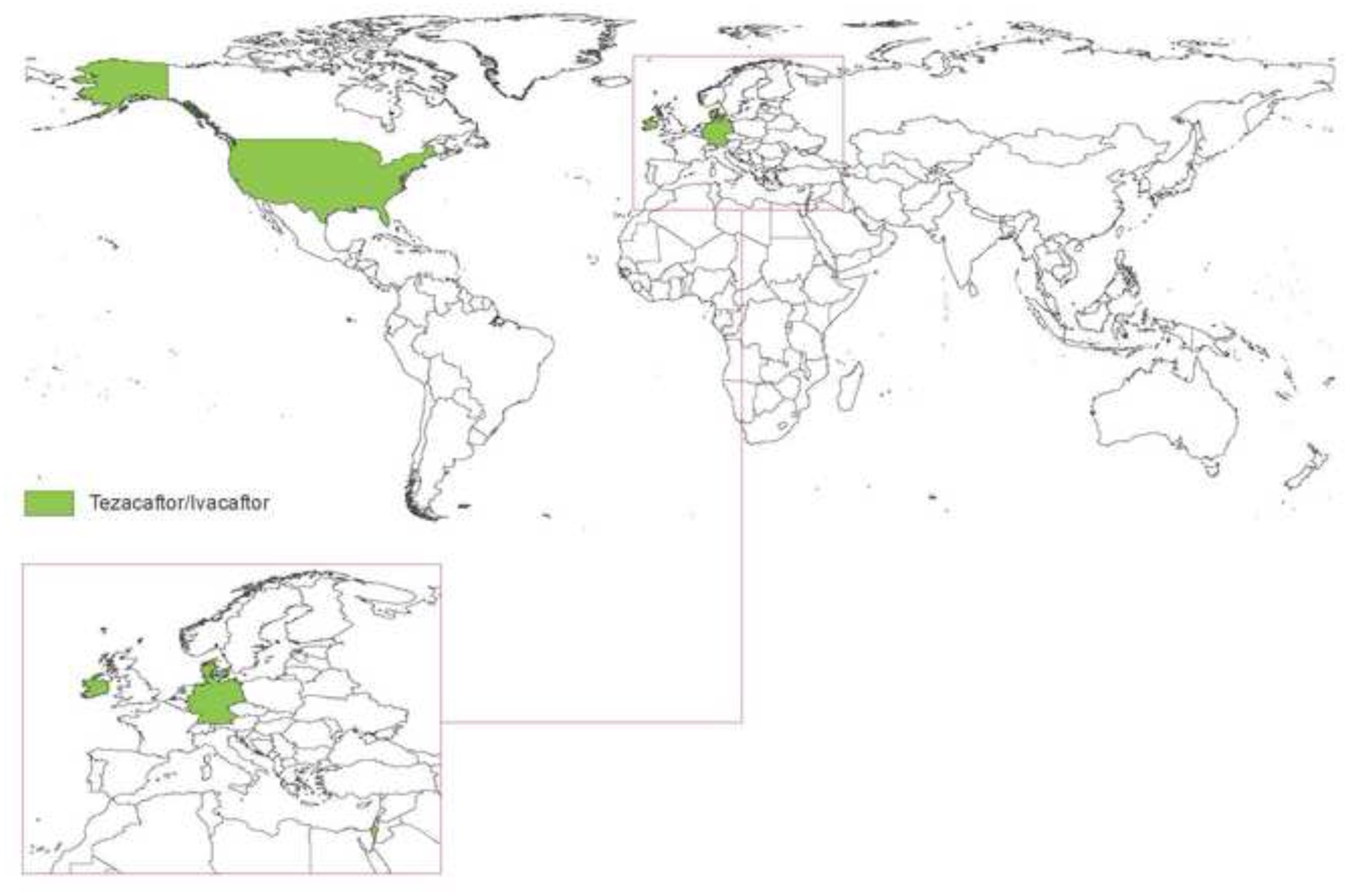
The past six decades have seen remarkable improvements in health outcomes for people with cystic fibrosis, which was once a fatal disease of infants and young children. However, although life expectancy for people with cystic fibrosis has increased substantially, the disease continues to limit survival and quality of life, and results in a large burden of care for people with cystic fibrosis and their families. Furthermore, epidemiological studies in the past two decades have shown that cystic fibrosis occurs and is more frequent than was previously thought in populations of non-European descent, and the disease is now recognised in many regions of the world. The Lancet Respiratory Medicine Commission on the future of cystic fibrosis care was established at a time of great change in the clinical care of people with the disease, with a growing population of adult patients, widespread genetic testing supporting the diagnosis of cystic fibrosis, and the development of therapies targeting defects in the cystic fibrosis transmembrane conductance regulator (CFTR), which are likely to affect the natural trajectory of the disease. The aim of the Commission was to bring to the attention of patients, health-care professionals, researchers, funders, service providers, and policy makers the various challenges associated with the changing landscape of cystic fibrosis care and the opportunities available for progress, providing a blueprint for the future of cystic fibrosis care. The discovery of the CFTR gene in the late 1980s triggered a surge of basic research that enhanced understanding of the pathophysiology and the genotype-phenotype relationships of this clinically variable disease. Until recently, available treatments could only control symptoms and restrict the complications of cystic fibrosis, but advances in CFTR modulator therapies to address the basic defect of cystic fibrosis have been remarkable and the field is evolving rapidly. However, CFTR modulators approved for use to date are highly expensive, which has prompted questions about the affordability of new treatments and served to emphasise the considerable gap in health outcomes for patients with cystic fibrosis between high-income countries, and low-income and middle-income countries (LMICs). Advances in clinical care have been multifaceted and include earlier diagnosis through the implementation of newborn screening programmes, formalised airway clearance therapy, and reduced malnutrition through the use of effective pancreatic enzyme replacement and a high-energy, high-protein diet. Centre-based care has become the norm in high-income countries, allowing patients to benefit from the skills of expert members of multidisciplinary teams. Pharmacological interventions to address respiratory manifestations now include drugs that target airway mucus and airway surface liquid hydration, and antimicrobial therapies such as antibiotic eradication treatment in early-stage infections and protocols for maintenance therapy of chronic infections. Despite the recent breakthrough with CFTR modulators for cystic fibrosis, the development of novel mucolytic, anti-inflammatory, and anti-infective therapies is likely to remain important, especially for patients with more advanced stages of lung disease. As the median age of patients with cystic fibrosis increases, with a rapid increase in the population of adults living with the disease, complications of cystic fibrosis are becoming increasingly common. Steps need to be taken to ensure that enough highly qualified professionals are present in cystic fibrosis centres to meet the needs of ageing patients, and new technologies need to be adopted to support communication between patients and health-care providers. In considering the future of cystic fibrosis care, the Commission focused on five key areas, which are discussed in this report: the changing epidemiology of cystic fibrosis (section 1); future challenges of clinical care and its delivery (section 2); the building of cystic fibrosis care globally (section 3); novel therapeutics (section 4); and patient engagement (section 5). In panel 1, we summarise key messages of the Commission. The challenges faced by all stakeholders in building and developing cystic fibrosis care globally are substantial, but many opportunities exist for improved care and health outcomes for patients in countries with established cystic fibrosis care programmes, and in LMICs where integrated multidisciplinary care is not available and resources are lacking at present. A concerted effort is needed to ensure that all patients with cystic fibrosis have access to high-quality health care in the future.
Copyright © 2020 Elsevier Ltd. All rights reserved.
Figures
Comment in
-
Cystic fibrosis lung disease and bronchiectasis.Lancet Respir Med. 2020 Jan;8(1):12-14. doi: 10.1016/S2213-2600(19)30335-2. Epub 2019 Sep 27. Lancet Respir Med. 2020. PMID: 31570316 No abstract available.
-
A patient's experience of cystic fibrosis care.Lancet Respir Med. 2020 Jan;8(1):14-16. doi: 10.1016/S2213-2600(19)30336-4. Epub 2019 Sep 27. Lancet Respir Med. 2020. PMID: 31570317 No abstract available.
-
Progress in understanding the molecular pathology and microbiology of cystic fibrosis.Lancet Respir Med. 2020 Jan;8(1):8-10. doi: 10.1016/S2213-2600(19)30333-9. Epub 2019 Sep 27. Lancet Respir Med. 2020. PMID: 31570319 No abstract available.
-
Clinical care for cystic fibrosis: preparing for the future now.Lancet Respir Med. 2020 Jan;8(1):10-12. doi: 10.1016/S2213-2600(19)30334-0. Epub 2019 Sep 27. Lancet Respir Med. 2020. PMID: 31570320 No abstract available.
-
Cystic fibrosis in Turkey.Lancet Respir Med. 2020 Apr;8(4):e17. doi: 10.1016/S2213-2600(20)30055-2. Lancet Respir Med. 2020. PMID: 32246929 No abstract available.
References
-
- Andersen DH, Hodges RG. Celiac syndrome; genetics of cystic fibrosis of the pancreas, with a consideration of etiology. Am J Dis Child 1946; 72: 62–80. - PubMed
-
- Di Sant’Agnese PA, Darling RC, Perera GA, Shea E. Abnormal electrolyte composition of sweat in cystic fibrosis of the pancreas; clinical significance and relationship to the disease. Pediatrics 1953; 12(5): 549–63. - PubMed
-
- Gibson LE, Cooke RE. A test for concentration of electrolytes in sweat in cystic fibrosis of the pancreas utilizing pilocarpine by iontophoresis. Pediatrics 1959; 23(3): 545–9. - PubMed
-
- Quinton PM. Chloride impermeability in cystic fibrosis. Nature 1983; 301(5899): 421–2. - PubMed
-
- Kerem B, Rommens JM, Buchanan JA, et al. Identification of the cystic fibrosis gene: genetic analysis. Science 1989; 245(4922): 1073–80. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
