

Whole-genome sequencing of triple-negative breast cancers in a population-based clinical study
- PMID: 31570822
- PMCID: PMC6859071
- DOI: 10.1038/s41591-019-0582-4
Whole-genome sequencing of triple-negative breast cancers in a population-based clinical study
Abstract
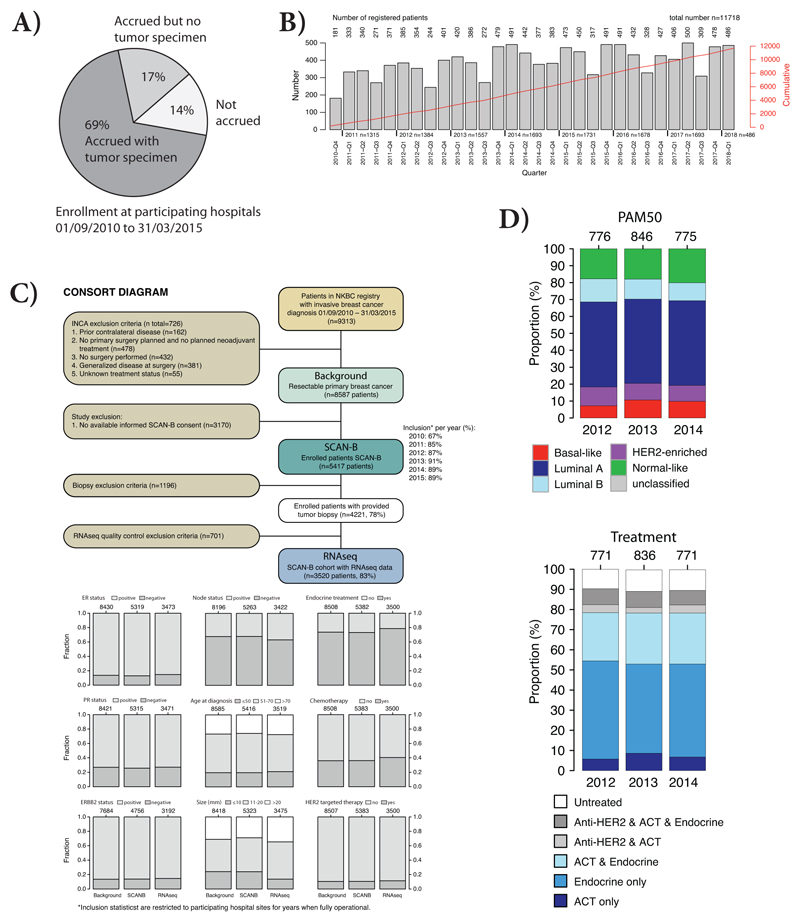
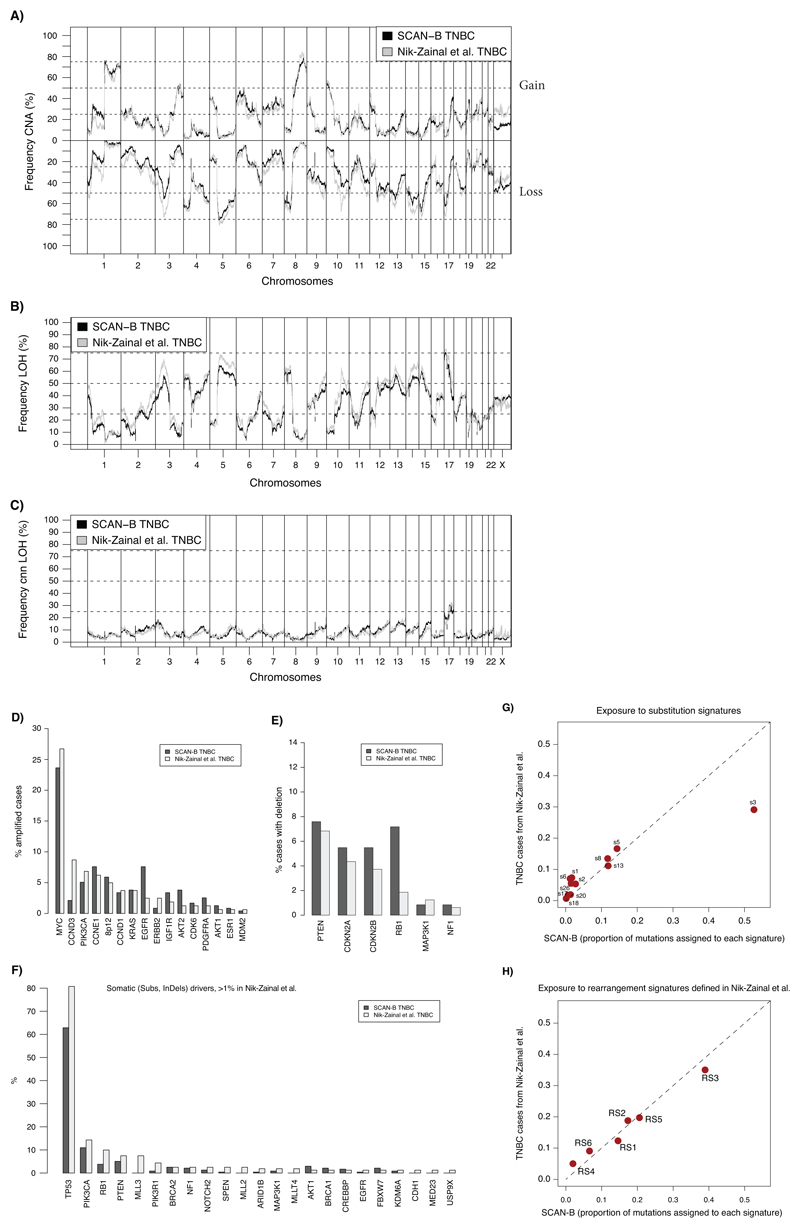
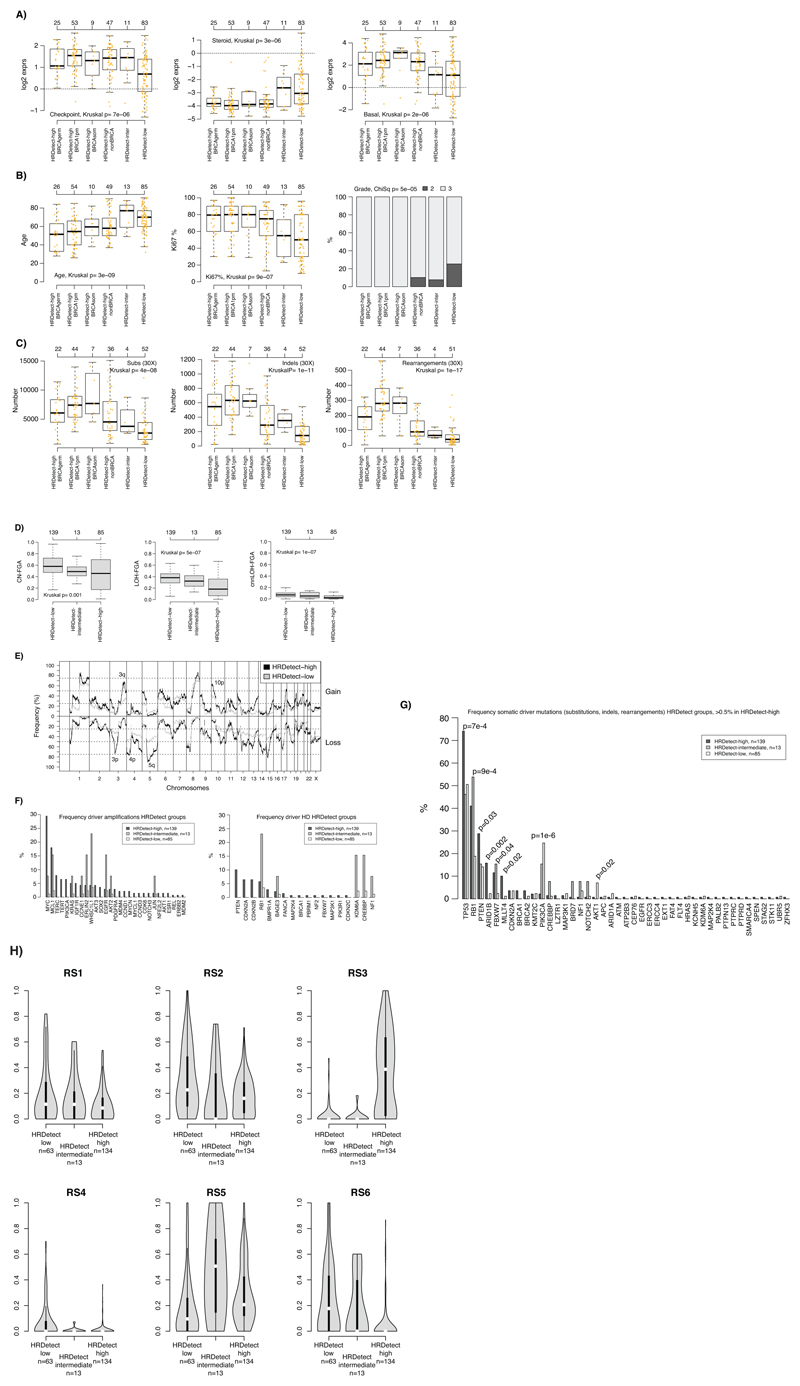
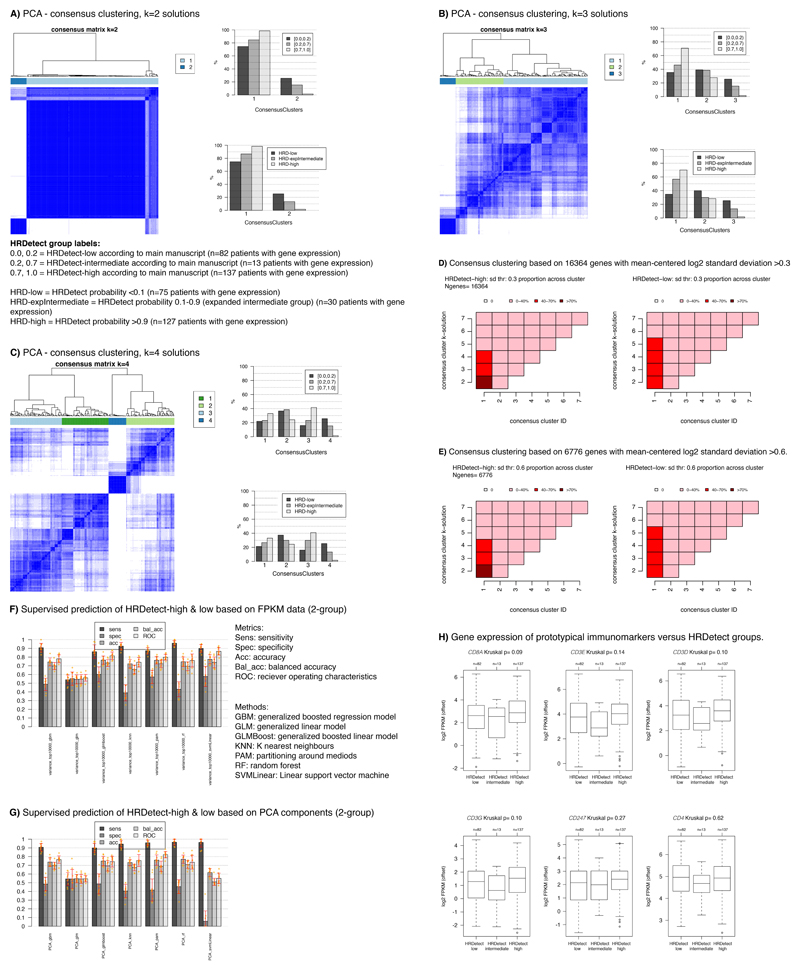
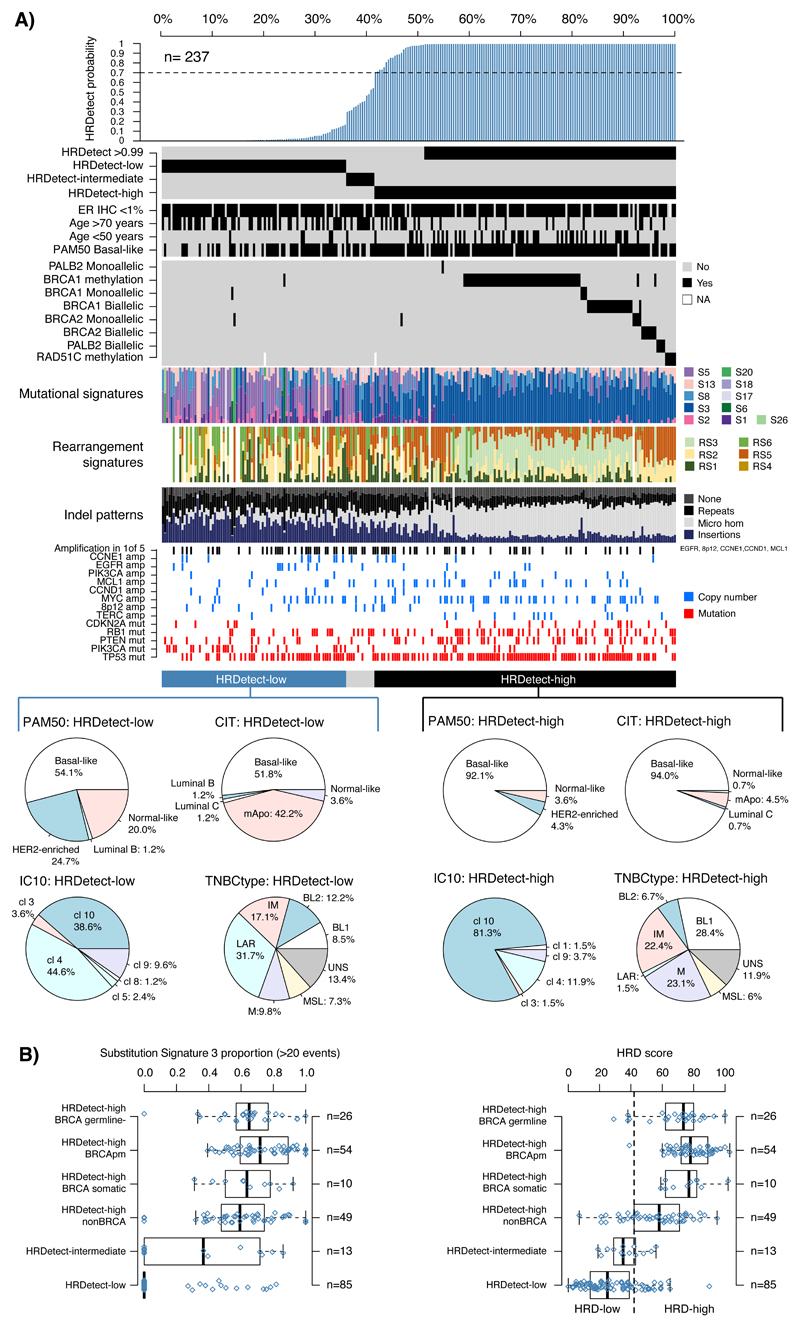
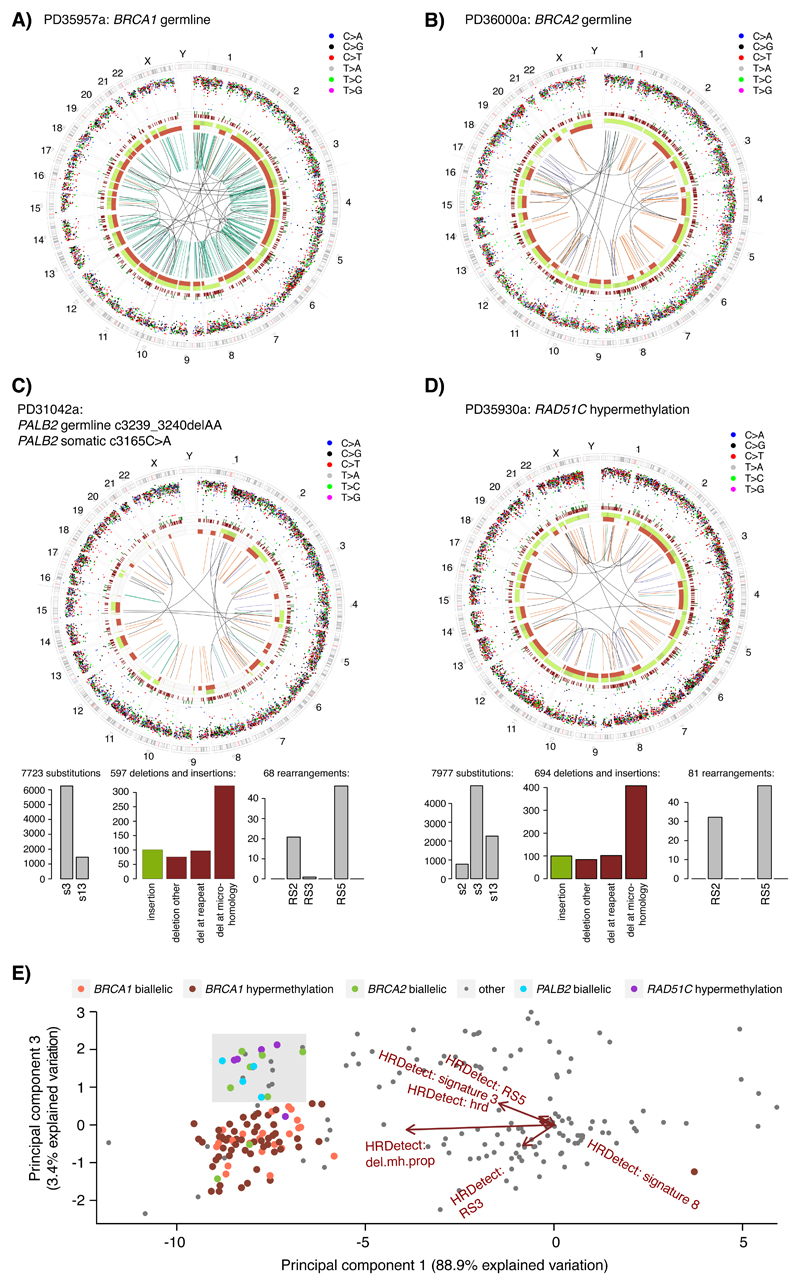
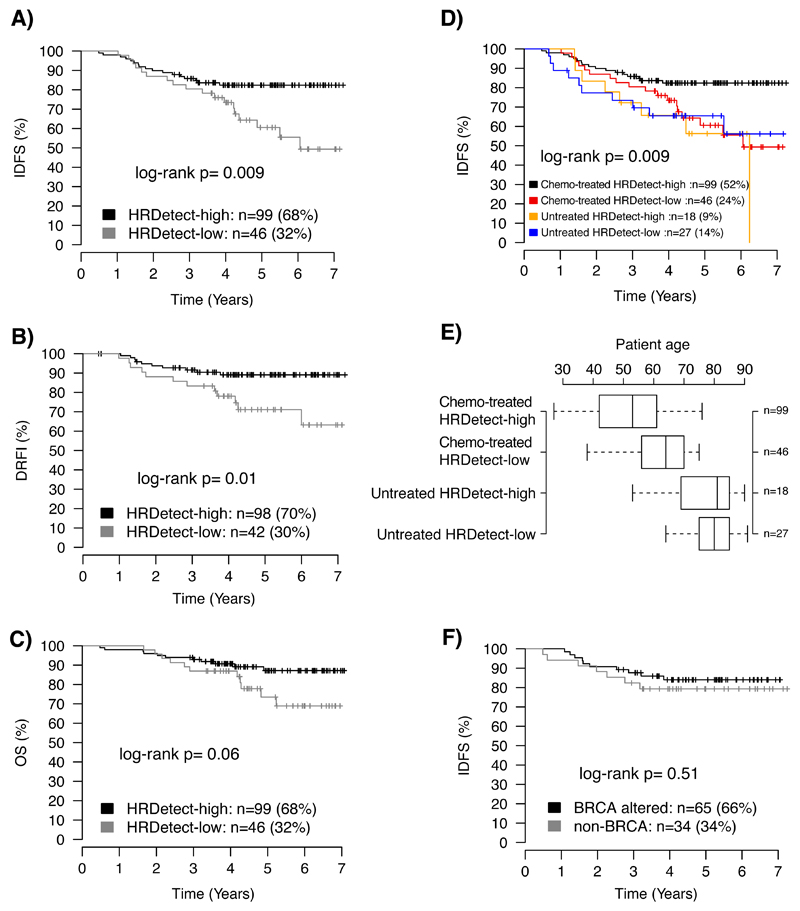
Whole-genome sequencing (WGS) brings comprehensive insights to cancer genome interpretation. To explore the clinical value of WGS, we sequenced 254 triple-negative breast cancers (TNBCs) for which associated treatment and outcome data were collected between 2010 and 2015 via the population-based Sweden Cancerome Analysis Network-Breast (SCAN-B) project (ClinicalTrials.gov ID:NCT02306096). Applying the HRDetect mutational-signature-based algorithm to classify tumors, 59% were predicted to have homologous-recombination-repair deficiency (HRDetect-high): 67% explained by germline/somatic mutations of BRCA1/BRCA2, BRCA1 promoter hypermethylation, RAD51C hypermethylation or biallelic loss of PALB2. A novel mechanism of BRCA1 abrogation was discovered via germline SINE-VNTR-Alu retrotransposition. HRDetect provided independent prognostic information, with HRDetect-high patients having better outcome on adjuvant chemotherapy for invasive disease-free survival (hazard ratio (HR) = 0.42; 95% confidence interval (CI) = 0.2-0.87) and distant relapse-free interval (HR = 0.31, CI = 0.13-0.76) compared to HRDetect-low, regardless of whether a genetic/epigenetic cause was identified. HRDetect-intermediate, some possessing potentially targetable biological abnormalities, had the poorest outcomes. HRDetect-low cancers also had inadequate outcomes: ~4.7% were mismatch-repair-deficient (another targetable defect, not typically sought) and they were enriched for (but not restricted to) PIK3CA/AKT1 pathway abnormalities. New treatment options need to be considered for now-discernible HRDetect-intermediate and HRDetect-low categories. This population-based study advocates for WGS of TNBC to better inform trial stratification and improve clinical decision-making.
Conflict of interest statement
D. Glodzik, H.R. Davies and S. Nik-Zainal are inventors on a patent encompassing the code and intellectual principle of the HRDetect algorithm. The remaining authors declare that they have no competing interests.
Figures
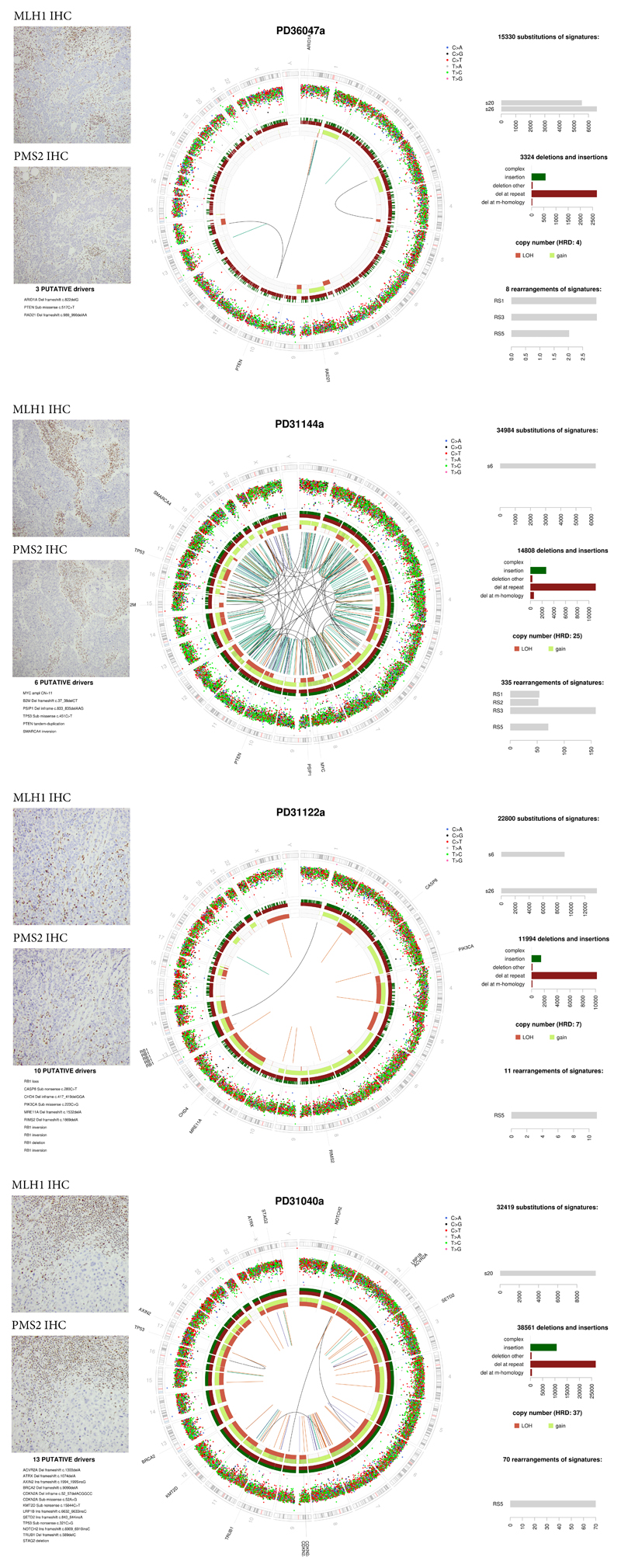
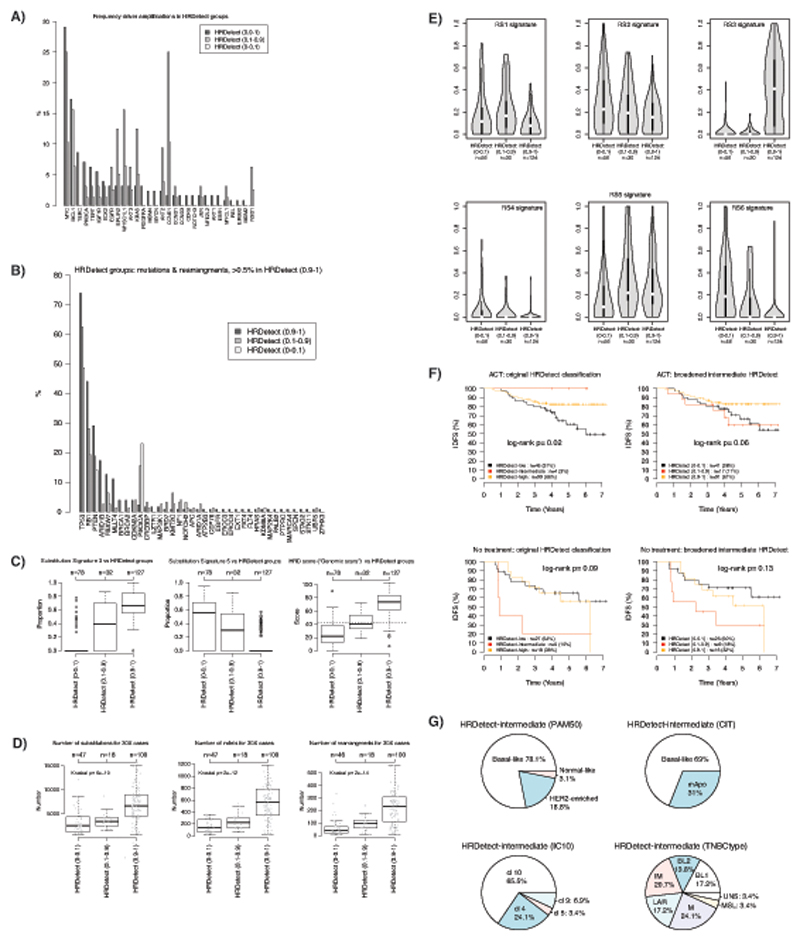
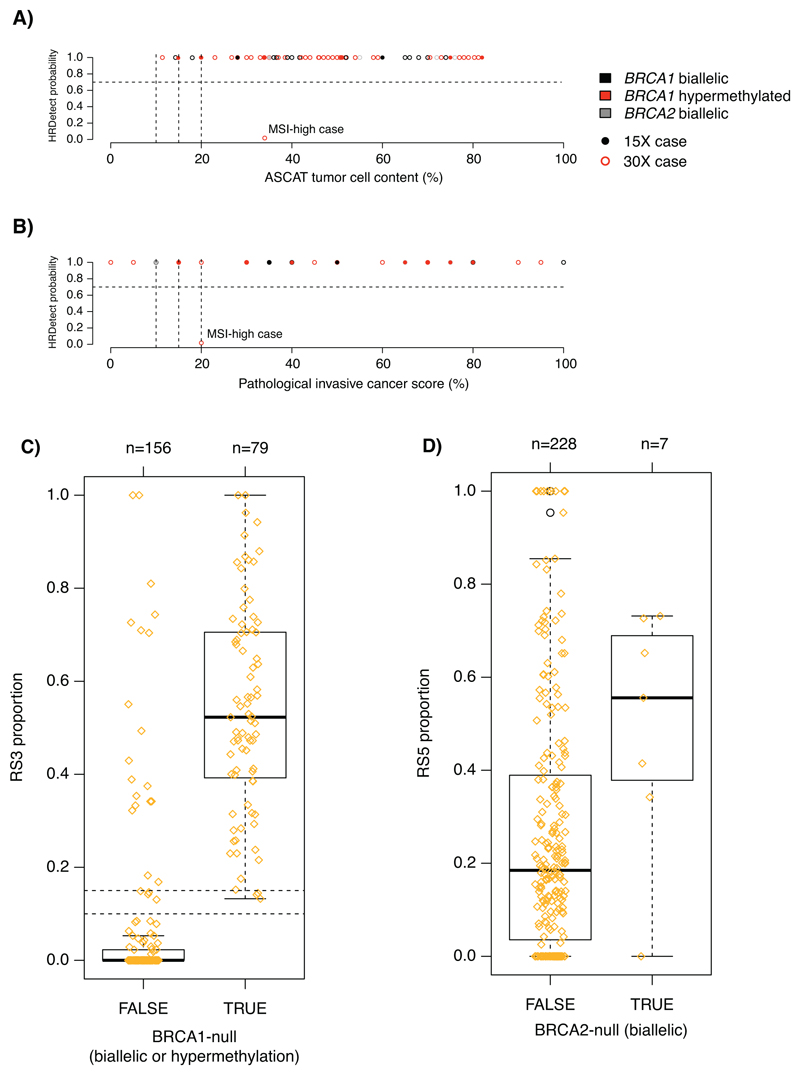
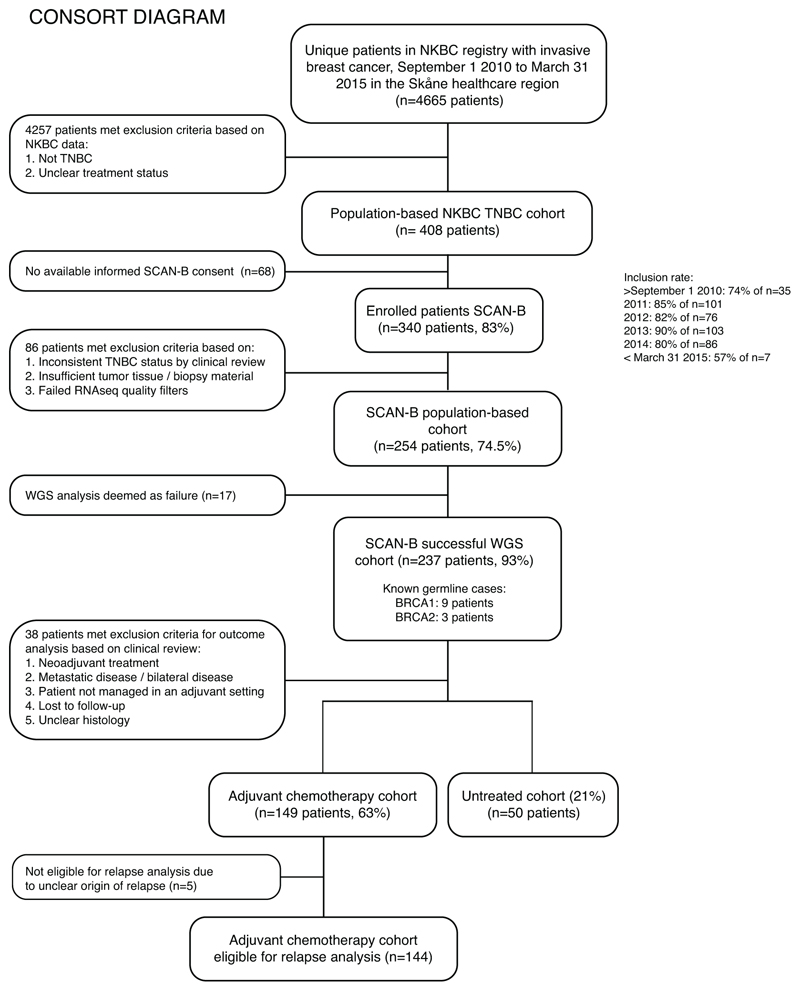
References
-
- Coe BP, et al. Resolving the resolution of array CGH. Genomics. 2007;89:647–653. - PubMed
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
