

Allele-specific NKX2-5 binding underlies multiple genetic associations with human electrocardiographic traits
- PMID: 31570892
- PMCID: PMC6858543
- DOI: 10.1038/s41588-019-0499-3
Allele-specific NKX2-5 binding underlies multiple genetic associations with human electrocardiographic traits
Abstract
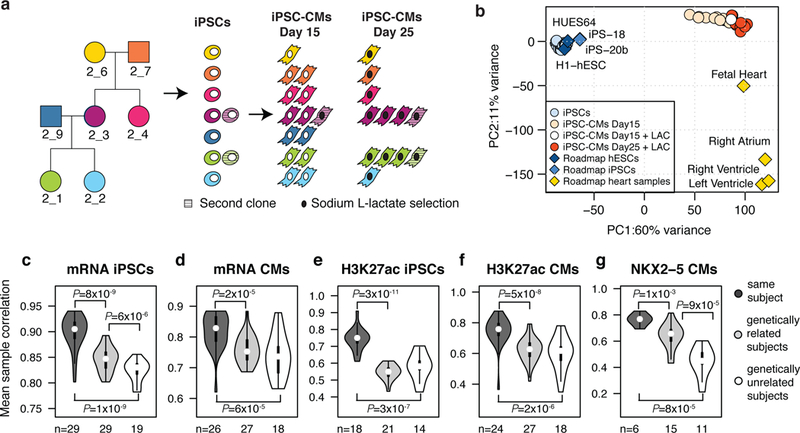
The cardiac transcription factor (TF) gene NKX2-5 has been associated with electrocardiographic (EKG) traits through genome-wide association studies (GWASs), but the extent to which differential binding of NKX2-5 at common regulatory variants contributes to these traits has not yet been studied. We analyzed transcriptomic and epigenomic data from induced pluripotent stem cell-derived cardiomyocytes from seven related individuals, and identified ~2,000 single-nucleotide variants associated with allele-specific effects (ASE-SNVs) on NKX2-5 binding. NKX2-5 ASE-SNVs were enriched for altered TF motifs, for heart-specific expression quantitative trait loci and for EKG GWAS signals. Using fine-mapping combined with epigenomic data from induced pluripotent stem cell-derived cardiomyocytes, we prioritized candidate causal variants for EKG traits, many of which were NKX2-5 ASE-SNVs. Experimentally characterizing two NKX2-5 ASE-SNVs (rs3807989 and rs590041) showed that they modulate the expression of target genes via differential protein binding in cardiac cells, indicating that they are functional variants underlying EKG GWAS signals. Our results show that differential NKX2-5 binding at numerous regulatory variants across the genome contributes to EKG phenotypes.
Figures






References
-
- Deplancke B, Alpern D & Gardeux V The genetics of transcription factor DNA binding variation. Cell 166, 538–554 (2016). - PubMed
Methods-only references
-
- Tohyama S et al. Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell 12, 127–137 (2013). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U01 DK105541/DK/NIDDK NIH HHS/United States
- R01 DK114650/DK/NIDDK NIH HHS/United States
- R37 DK039949/DK/NIDDK NIH HHS/United States
- DP3 DK112155/DK/NIDDK NIH HHS/United States
- R41 HG008118/HG/NHGRI NIH HHS/United States
- U01 HL107442/HL/NHLBI NIH HHS/United States
- F31 HL142151/HL/NHLBI NIH HHS/United States
- 155105/SNSF_/Swiss National Science Foundation/Switzerland
- R01 DK018477/DK/NIDDK NIH HHS/United States
- P30 CA023100/CA/NCI NIH HHS/United States
- R01 DK039949/DK/NIDDK NIH HHS/United States
- R44 HG008118/HG/NHGRI NIH HHS/United States
- T32 GM008666/GM/NIGMS NIH HHS/United States
- 167612/SNSF_/Swiss National Science Foundation/Switzerland
- R01 HL141989/HL/NHLBI NIH HHS/United States
- R01 HL116747/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
