

Landscape of stimulation-responsive chromatin across diverse human immune cells
- PMID: 31570894
- PMCID: PMC6858557
- DOI: 10.1038/s41588-019-0505-9
Landscape of stimulation-responsive chromatin across diverse human immune cells
Abstract
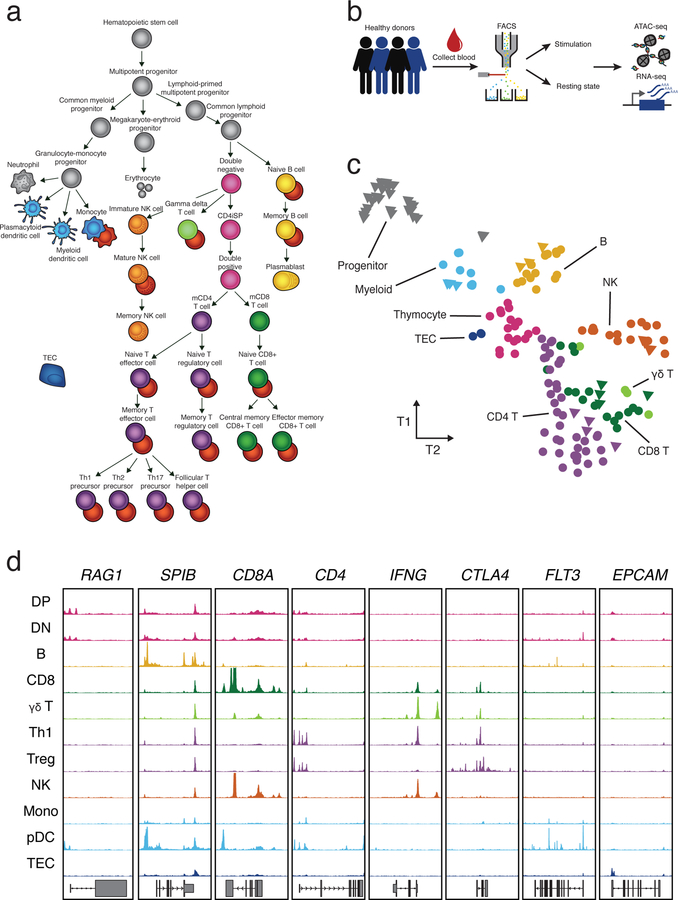
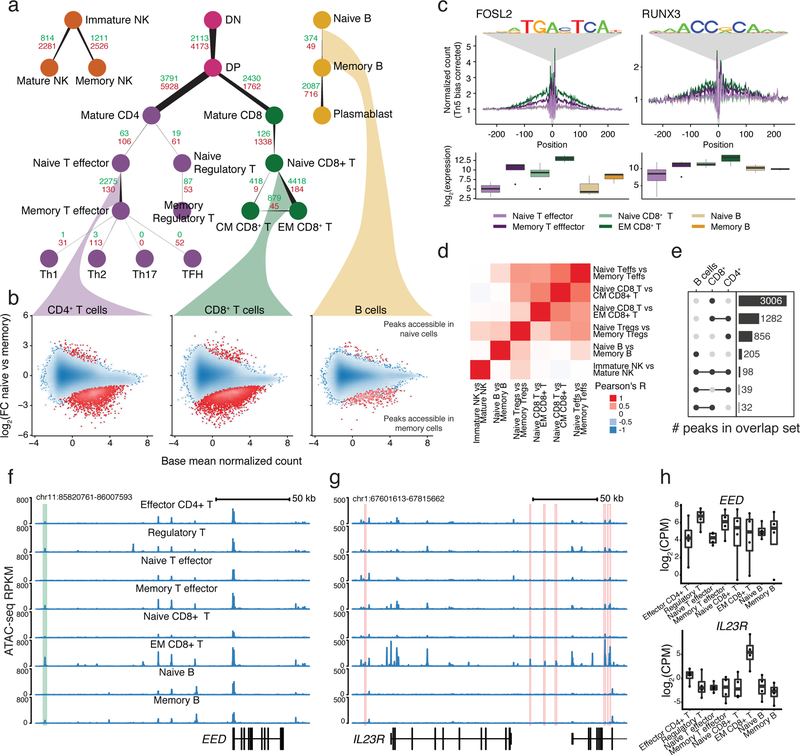
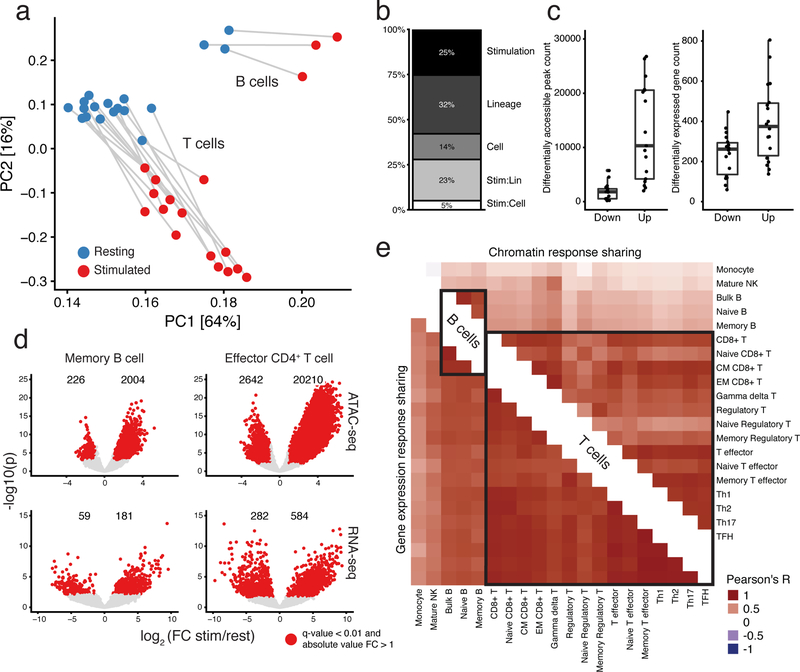
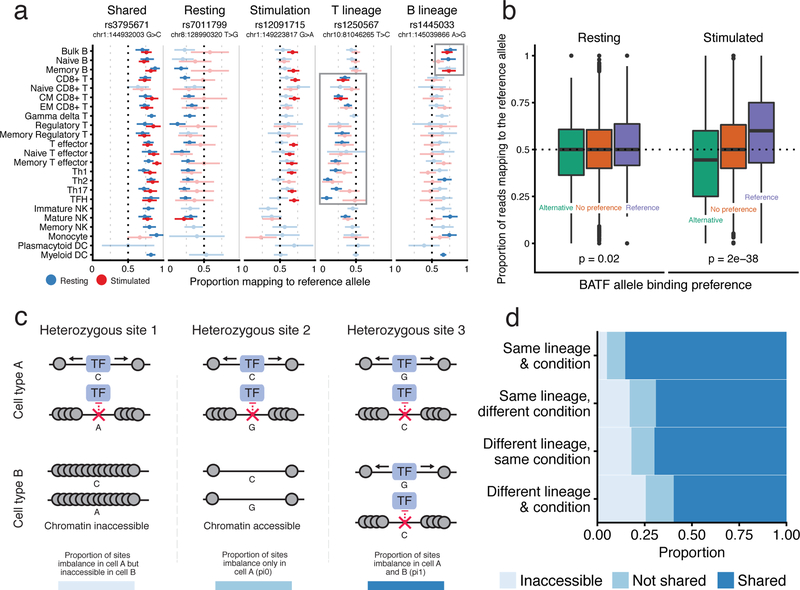
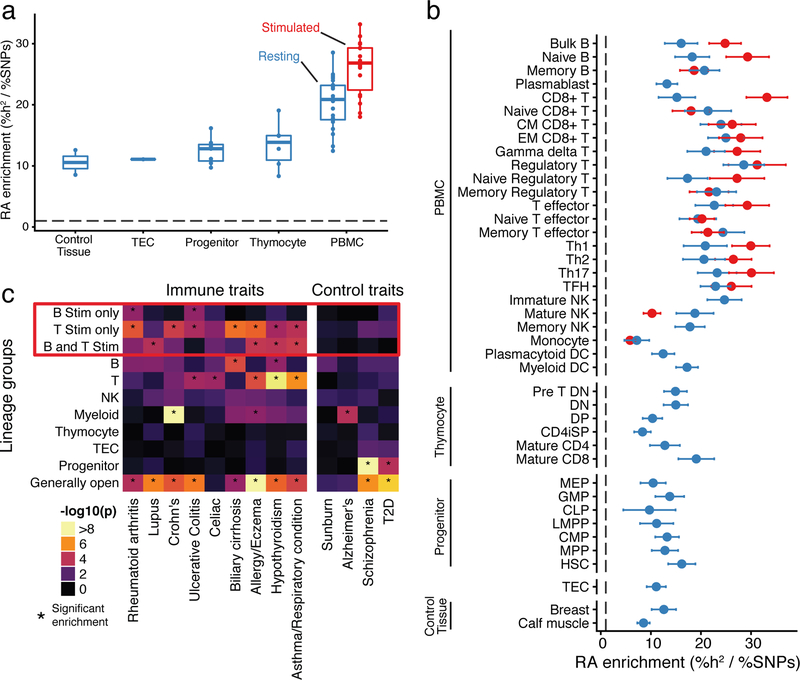
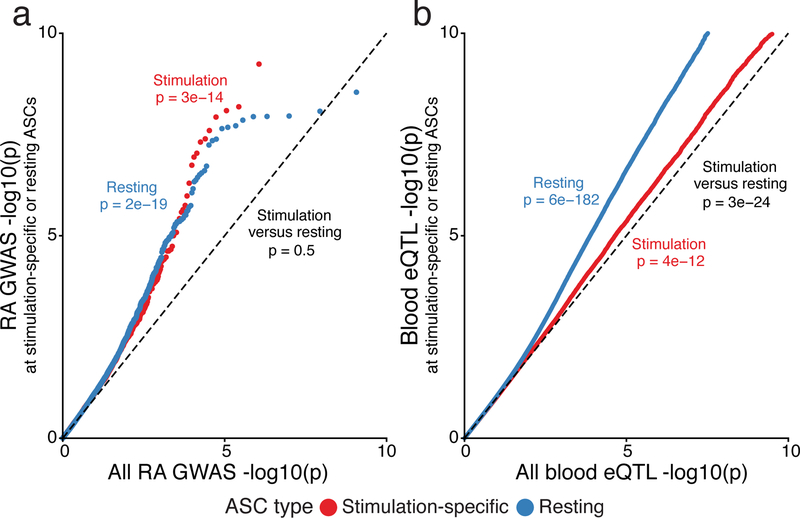
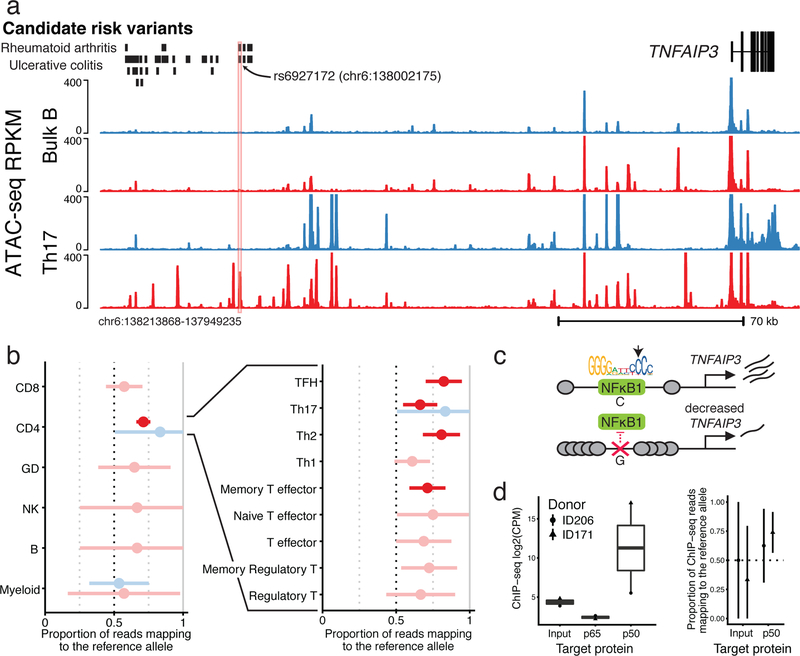
A hallmark of the immune system is the interplay among specialized cell types transitioning between resting and stimulated states. The gene regulatory landscape of this dynamic system has not been fully characterized in human cells. Here we collected assay for transposase-accessible chromatin using sequencing (ATAC-seq) and RNA sequencing data under resting and stimulated conditions for up to 32 immune cell populations. Stimulation caused widespread chromatin remodeling, including response elements shared between stimulated B and T cells. Furthermore, several autoimmune traits showed significant heritability in stimulation-responsive elements from distinct cell types, highlighting the importance of these cell states in autoimmunity. Allele-specific read mapping identified variants that alter chromatin accessibility in particular conditions, allowing us to observe evidence of function for a candidate causal variant that is undetected by existing large-scale studies in resting cells. Our results provide a resource of chromatin dynamics and highlight the need to characterize the effects of genetic variation in stimulated cells.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- RM1 HG007735/HG/NHGRI NIH HHS/United States
- R01 HG008140/HG/NHGRI NIH HHS/United States
- U19 AI057266/AI/NIAID NIH HHS/United States
- S10 OD021822/OD/NIH HHS/United States
- P30 AR070155/AR/NIAMS NIH HHS/United States
- P50 HG007735/HG/NHGRI NIH HHS/United States
- T15 LM007033/LM/NLM NIH HHS/United States
- P50 GM082250/GM/NIGMS NIH HHS/United States
- UM1 HG009442/HG/NHGRI NIH HHS/United States
- P50 AI150476/AI/NIAID NIH HHS/United States
- S10 OD018220/OD/NIH HHS/United States
- P30 DK063720/DK/NIDDK NIH HHS/United States
- DP3 DK111914/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
