

Probiotic Lactobacillus rhamnosus GG Prevents Liver Fibrosis Through Inhibiting Hepatic Bile Acid Synthesis and Enhancing Bile Acid Excretion in Mice
- PMID: 31571251
- PMCID: PMC7317518
- DOI: 10.1002/hep.30975
Probiotic Lactobacillus rhamnosus GG Prevents Liver Fibrosis Through Inhibiting Hepatic Bile Acid Synthesis and Enhancing Bile Acid Excretion in Mice
Abstract
Background and aims: Cholestatic liver disease is characterized by gut dysbiosis and excessive toxic hepatic bile acids (BAs). Modification of gut microbiota and repression of BA synthesis are potential strategies for the treatment of cholestatic liver disease. The purpose of this study was to examine the effects and to understand the mechanisms of the probiotic Lactobacillus rhamnosus GG (LGG) on hepatic BA synthesis, liver injury, and fibrosis in bile duct ligation (BDL) and multidrug resistance protein 2 knockout (Mdr2-/- ) mice.
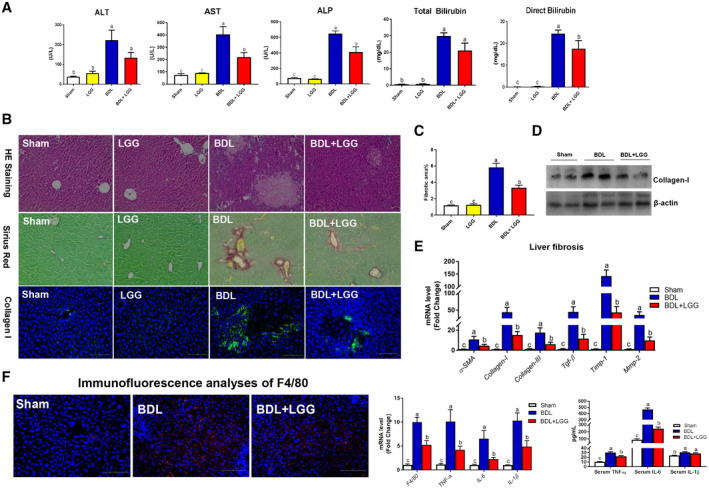
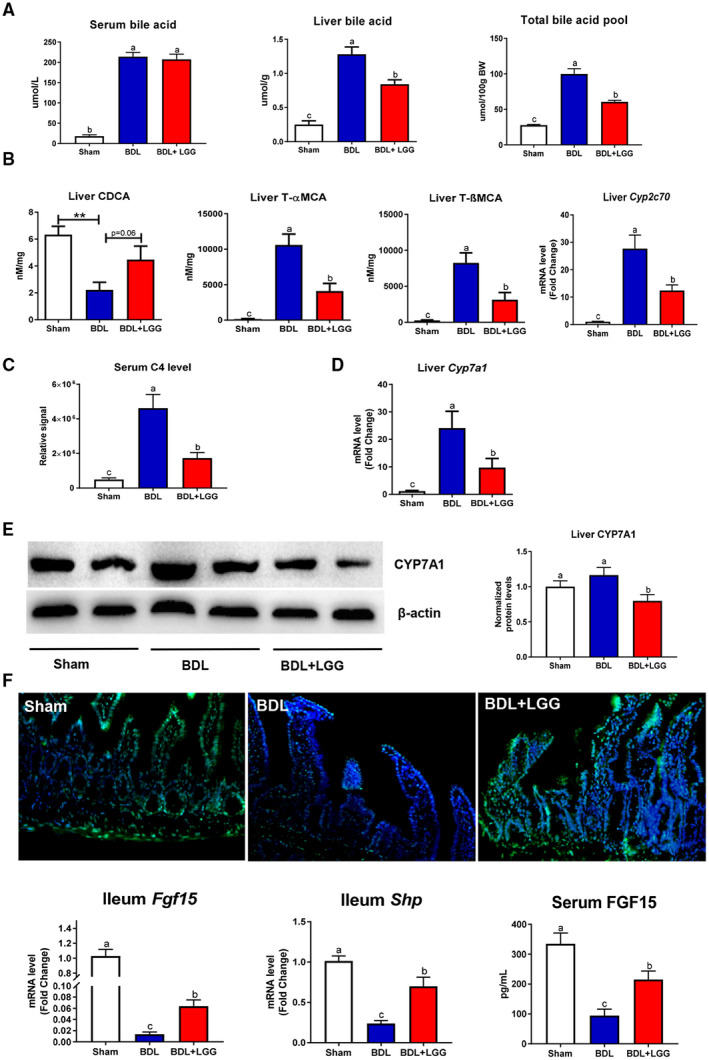
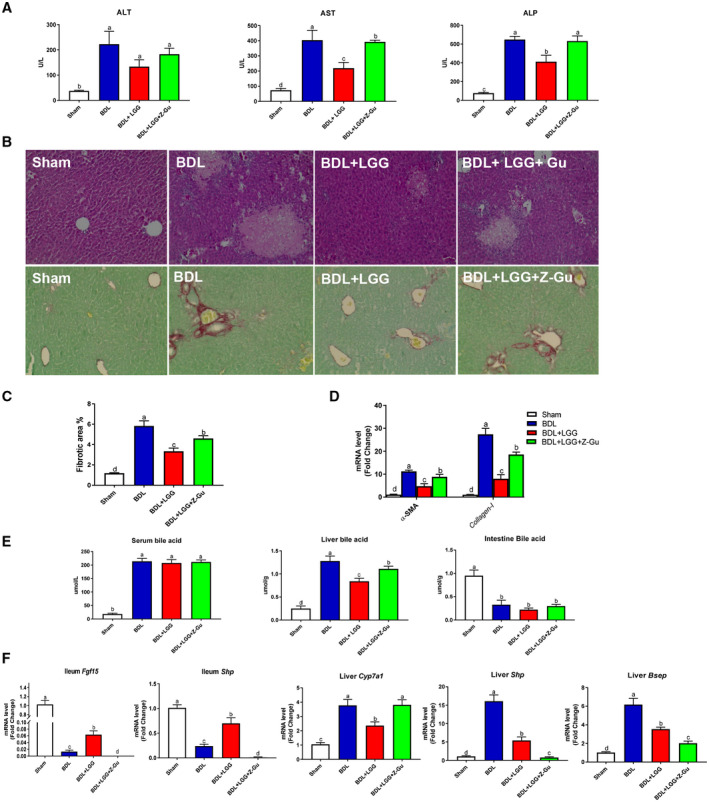
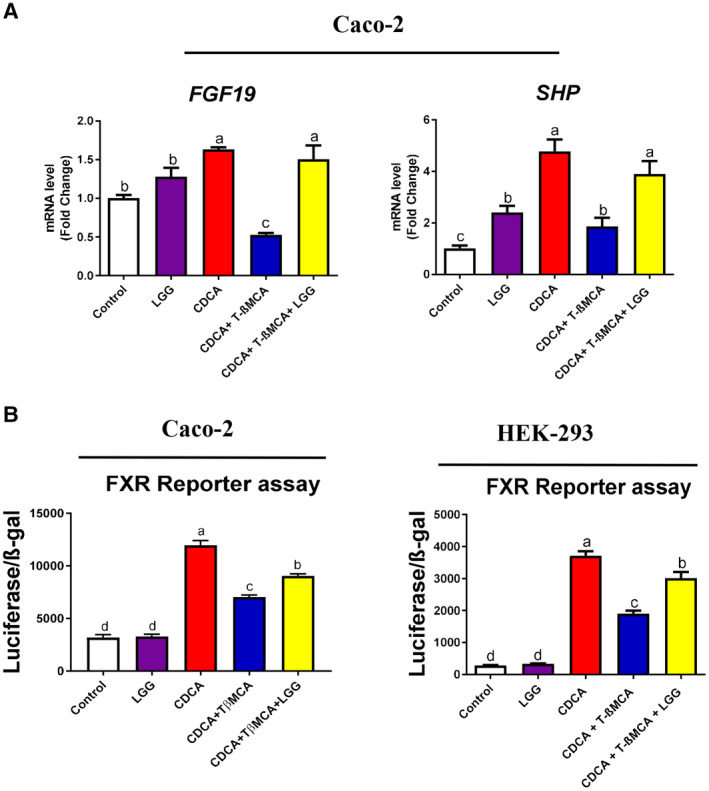
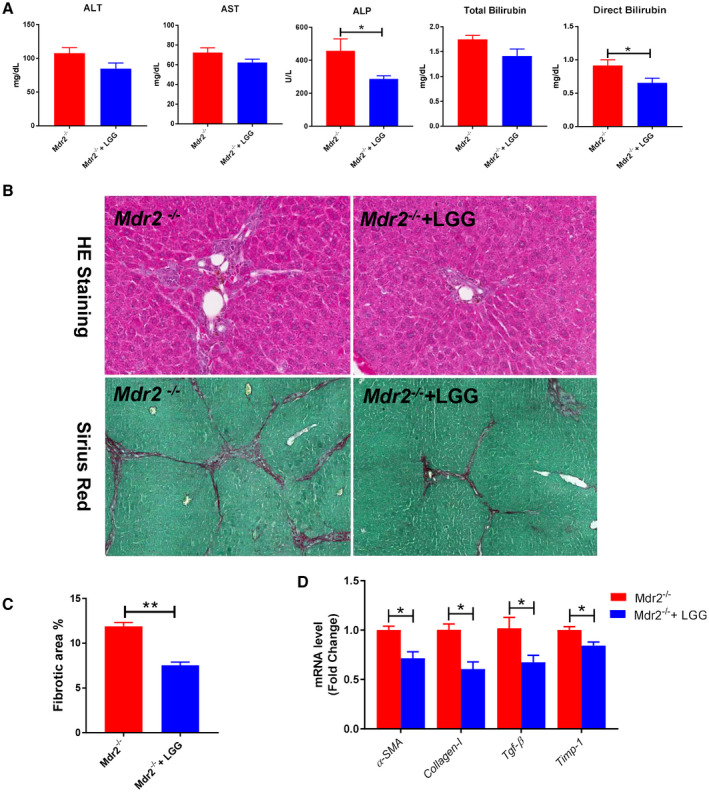
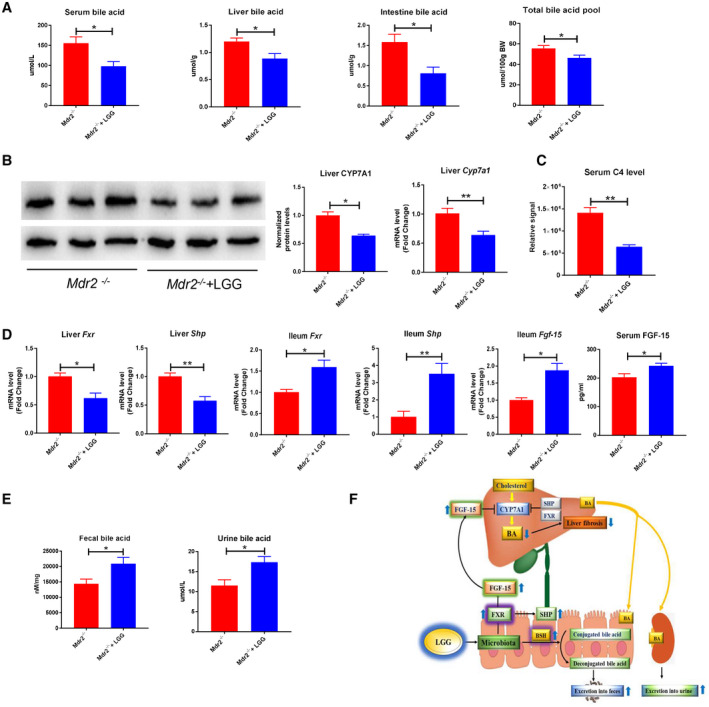
Approach and results: Global and intestine-specific farnesoid X receptor (FXR) inhibitors were used to dissect the role of FXR. LGG treatment significantly attenuated liver inflammation, injury, and fibrosis with a significant reduction of hepatic BAs in BDL mice. Hepatic concentration of taurine-β-muricholic acid (T-βMCA), an FXR antagonist, was markedly increased in BDL mice and reduced in LGG-treated mice, while chenodeoxycholic acid, an FXR agonist, was decreased in BDL mice and normalized in LGG-treated mice. LGG treatment significantly increased the expression of serum and ileum fibroblast growth factor 15 (FGF-15) and subsequently reduced hepatic cholesterol 7α-hydroxylase and BA synthesis in BDL and Mdr2-/- mice. At the molecular level, these changes were reversed by global and intestine-specific FXR inhibitors in BDL mice. In addition, LGG treatment altered gut microbiota, which was associated with increased BA deconjugation and increased fecal and urine BA excretion in both BDL and Mdr2-/- mice. In vitro studies showed that LGG suppressed the inhibitory effect of T-βMCA on FXR and FGF-19 expression in Caco-2 cells.
Conclusion: LGG supplementation decreases hepatic BA by increasing intestinal FXR-FGF-15 signaling pathway-mediated suppression of BA de novo synthesis and enhances BA excretion, which prevents excessive BA-induced liver injury and fibrosis in mice.
© 2019 The Authors. Hepatology published by Wiley Periodicals, Inc., on behalf of American Association for the Study of Liver Diseases.
Figures


Comment in
-
Fine tuning the gut-liver-axis.Arch Toxicol. 2020 Oct;94(10):3595-3596. doi: 10.1007/s00204-020-02886-0. Epub 2020 Sep 5. Arch Toxicol. 2020. PMID: 32889577 No abstract available.
References
-
- Jansen PL, Ghallab A, Vartak N, Reif R, Schaap FG, Hampe J, et al. The ascending pathophysiology of cholestatic liver disease. Hepatology 2017;65:722‐738. - PubMed
-
- Schaap FG, Trauner M, Jansen PL. Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol 2014;11:55‐67. - PubMed
-
- Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab 2005;2:217‐225. - PubMed
-
- Modica S, Petruzzelli M, Bellafante E, Murzilli S, Salvatore L, Celli N, et al. Selective activation of nuclear bile acid receptor FXR in the intestine protects mice against cholestasis. Gastroenterology 2012;142:355‐365. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P50AA024337/AA/NIAAA NIH HHS/United States
- P20 GM113226/GM/NIGMS NIH HHS/United States
- U01AA022489-01A1/AA/NIAAA NIH HHS/United States
- R01AA023681/AA/NIAAA NIH HHS/United States
- R21 AA020848/AA/NIAAA NIH HHS/United States
- U01AA021893-01/AA/NIAAA NIH HHS/United States
- 1I01BX002996/VA/VA/United States
- R01AA023190/AA/NIAAA NIH HHS/United States
- R01 AA023190/AA/NIAAA NIH HHS/United States
- R01 AA023681/AA/NIAAA NIH HHS/United States
- U01 AA022489/AA/NIAAA NIH HHS/United States
- R21 AA022416/AA/NIAAA NIH HHS/United States
- I01 BX002996/BX/BLRD VA/United States
- U01 AA021893/AA/NIAAA NIH HHS/United States
- U01 AA021901/AA/NIAAA NIH HHS/United States
- U01AA021901/AA/NIAAA NIH HHS/United States
- P20GM113226/GM/NIGMS NIH HHS/United States
- P50 AA024337/AA/NIAAA NIH HHS/United States
- R21AA020848/AA/NIAAA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
