

Amplified fragment length polymorphism and whole genome sequencing: a comparison of methods in the investigation of a nosocomial outbreak with vancomycin resistant enterococci
- PMID: 31572571
- PMCID: PMC6757385
- DOI: 10.1186/s13756-019-0604-5
Amplified fragment length polymorphism and whole genome sequencing: a comparison of methods in the investigation of a nosocomial outbreak with vancomycin resistant enterococci
Abstract
Background: Recognition of nosocomial outbreaks with antimicrobial resistant (AMR) pathogens and appropriate infection prevention measures are essential to limit the consequences of AMR pathogens to patients in hospitals. Because unrelated, but genetically similar AMR pathogens may circulate simultaneously, rapid high-resolution molecular typing methods are needed for outbreak management. We compared amplified fragment length polymorphism (AFLP) and whole genome sequencing (WGS) during a nosocomial outbreak of vancomycin-resistant Enterococcus faecium (VRE) that spanned 5 months.
Methods: Hierarchical clustering of AFLP profiles was performed using unweighted pair-grouping and similarity coefficients were calculated with Pearson correlation. For WGS-analysis, core single nucleotide polymorphisms (SNPs) were used to calculate the pairwise distance between isolates, construct a maximum likelihood phylogeny and establish a cut-off for relatedness of epidemiologically linked VRE isolates. SNP-variations in the vanB gene cluster were compared to increase the comparative resolution. Technical replicates of 2 isolates were sequenced to determine the number of core-SNPs derived from random sequencing errors.
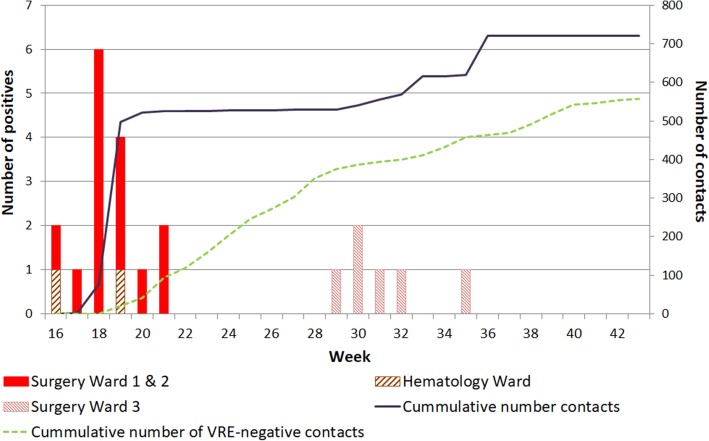
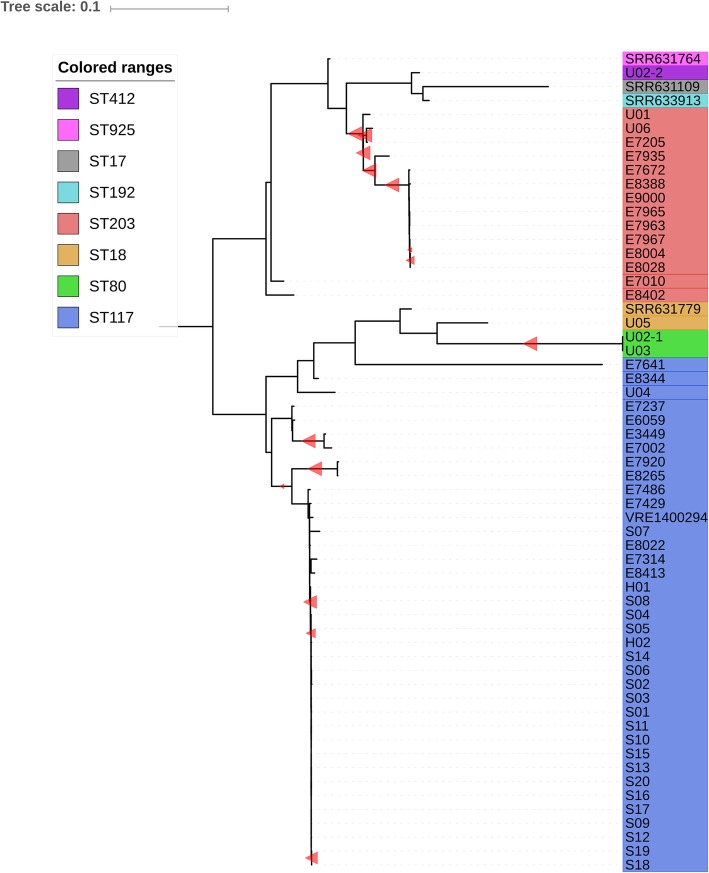
Results: Of the 721 patients screened for VRE carriage, AFLP assigned isolates of 22 patients to the outbreak cluster. According to WGS, all 22 isolates belonged to ST117 but only 21 grouped in a tight phylogenetic cluster and carried vanB resistance gene clusters. Sequencing of technical replicates showed that 4-5 core-SNPs were derived by random sequencing errors. The cut-off for relatedness of epidemiologically linked VRE isolates was established at ≤7 core-SNPs. The discrepant isolate was separated from the index isolate by 61 core-SNPs and the vanB gene cluster was absent. In AFLP analysis this discrepant isolate was indistinguishable from the other outbreak isolates, forming a cluster with 92% similarity (cut-off for identical isolates ≥90%). The inclusion of the discrepant isolate in the outbreak resulted in the screening of 250 patients and quarantining of an entire ward.
Conclusion: AFLP was a rapid and affordable screening tool for characterising hospital VRE outbreaks. For in-depth understanding of the outbreak WGS was needed. Compared to AFLP, WGS provided higher resolution typing of VRE isolates with implications for outbreak management.
Keywords: AFLP; Molecular typing; Nosocomial outbreak; VRE; WGS.
© The Author(s). 2019.
Conflict of interest statement
Competing interestsThe authors declare that they have no competing interests.
Figures

References
-
- Cassini A, Högberg LD, Plachouras D, Quattrocchi A, Hoxha A, Simonsen GS, et al. Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: a population-level modelling analysis. Lancet Infect Dis. 2018;0(0) Available from: http://www.ncbi.nlm.nih.gov/pubmed/30409683. Cited 7 Dec 2018. - PMC - PubMed
-
- van Hal SJ, Ip CLC, Ansari MA, Wilson DJ, Espedido BA, Jensen SO, et al. Evolutionary dynamics of Enterococcus faecium reveals complex genomic relationships between isolates with independent emergence of vancomycin resistance. Microb Genomics. 2016;2(1). Available from: 10.1099/mgen.0.000048. Cited 29 Nov 2018. - PMC - PubMed
-
- Veenemans J, Overdevest IT, Snelders E, Willemsen I, Hendriks Y, Adesokan A, et al. Next-generation sequencing for typing and detection of resistance genes: performance of a new commercial method during an outbreak of extended-spectrum-beta-lactamase-producing Escherichia coli. J Clin Microbiol. 2014;52(7):2454–2460. doi: 10.1128/JCM.00313-14. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
