

Addressing Patient Specificity in the Engineering of Tumor Models
- PMID: 31572718
- PMCID: PMC6751285
- DOI: 10.3389/fbioe.2019.00217
Addressing Patient Specificity in the Engineering of Tumor Models
Abstract
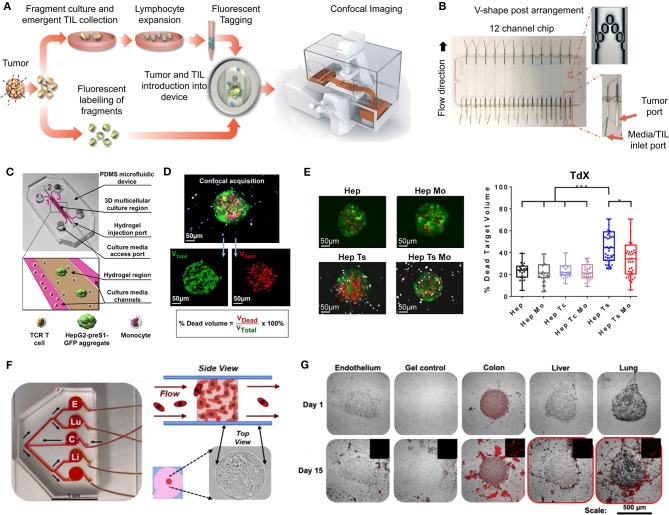
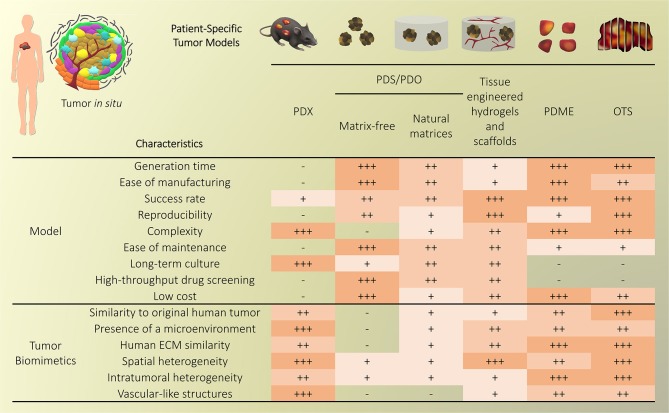
Cancer treatment is challenged by the heterogeneous nature of cancer, where prognosis depends on tumor type and disease stage, as well as previous treatments. Optimal patient stratification is critical for the development and validation of effective treatments, yet pre-clinical model systems are lacking in the delivery of effective individualized platforms that reflect distinct patient-specific clinical situations. Advances in cancer cell biology, biofabrication, and microengineering technologies have led to the development of more complex in vitro three-dimensional (3D) models to act as drug testing platforms and to elucidate novel cancer mechanisms. Mostly, these strategies have enabled researchers to account for the tumor microenvironment context including tumor-stroma interactions, a key factor of heterogeneity that affects both progression and therapeutic resistance. This is aided by state-of-the-art biomaterials and tissue engineering technologies, coupled with reproducible and high-throughput platforms that enable modeling of relevant physical and chemical factors. Yet, the translation of these models and technologies has been impaired by neglecting to incorporate patient-derived cells or tissues, and largely focusing on immortalized cell lines instead, contributing to drug failure rates. While this is a necessary step to establish and validate new models, a paradigm shift is needed to enable the systematic inclusion of patient-derived materials in the design and use of such models. In this review, we first present an overview of the components responsible for heterogeneity in different tumor microenvironments. Next, we introduce the state-of-the-art of current in vitro 3D cancer models employing patient-derived materials in traditional scaffold-free approaches, followed by novel bioengineered scaffold-based approaches, and further supported by dynamic systems such as bioreactors, microfluidics, and tumor-on-a-chip devices. We critically discuss the challenges and clinical prospects of models that have succeeded in providing clinical relevance and impact, and present emerging concepts of novel cancer model systems that are addressing patient specificity, the next frontier to be tackled by the field.
Keywords: 3D tumor models; hydrogels; microfluidics; patient-derived; primary cells; tissue engineering; tumor heterogeneity; tumor microenvironment.
Copyright © 2019 Bray, Hutmacher and Bock.
Figures

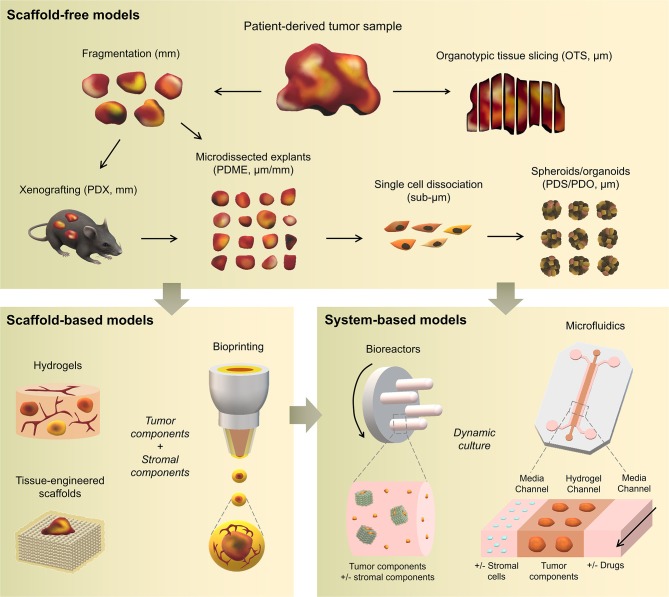


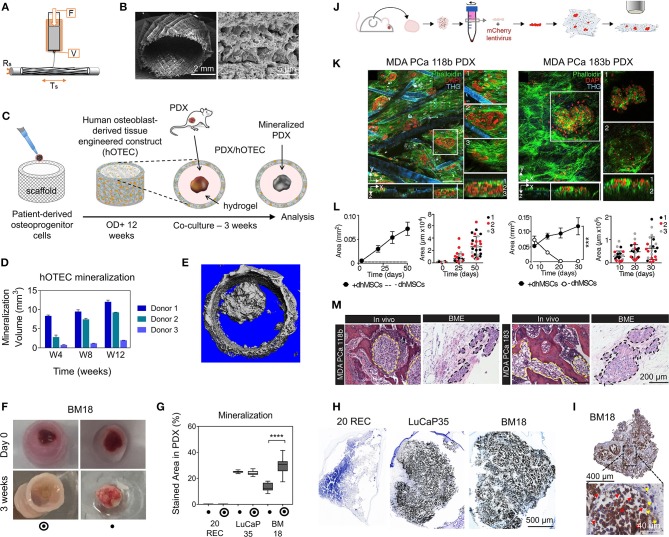
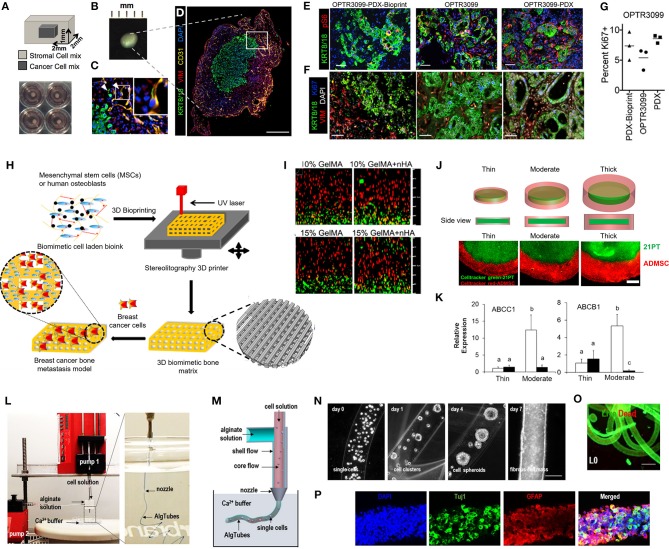

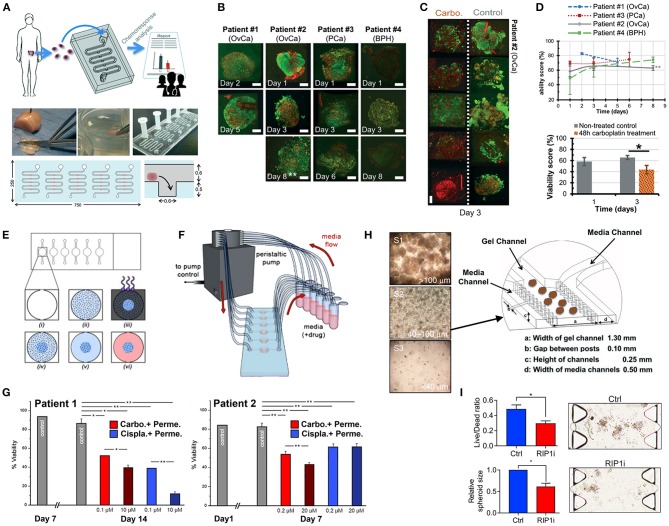
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources