

Modification of the PROM1 disease phenotype by a mutation in ABCA4
- PMID: 31576780
- PMCID: PMC6777736
- DOI: 10.1080/13816810.2019.1660382
Modification of the PROM1 disease phenotype by a mutation in ABCA4
Abstract
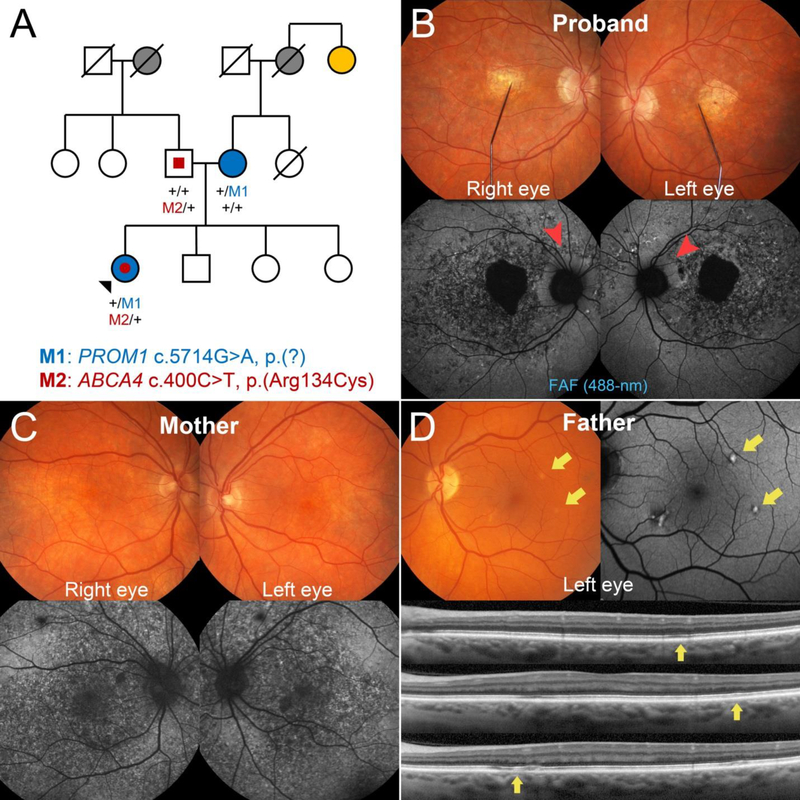
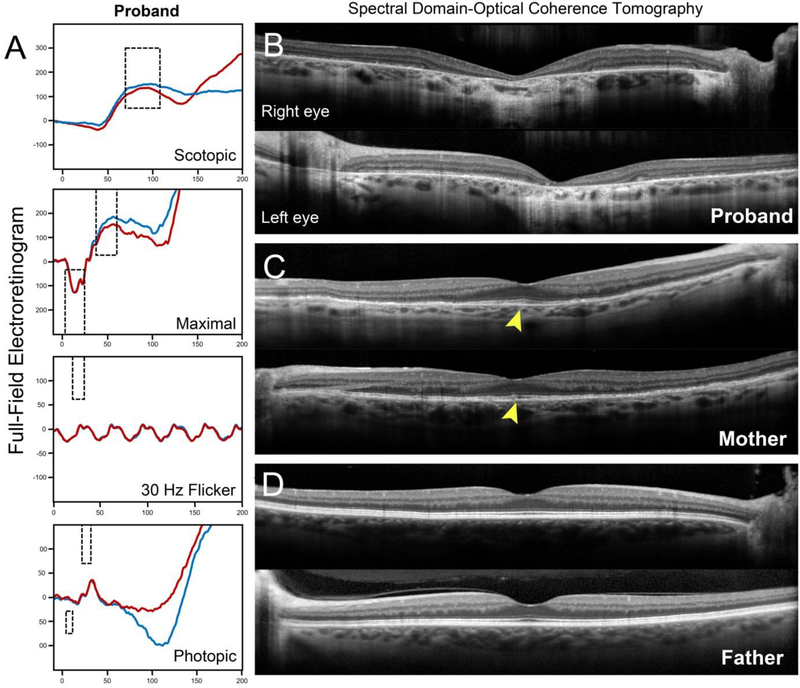
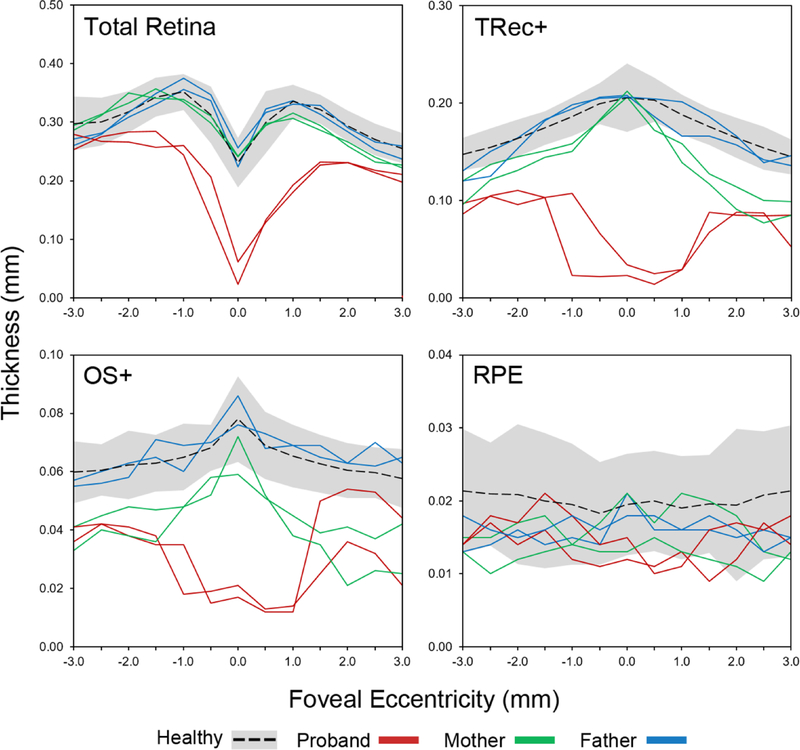
Background: The extensive phenotypic heterogeneity of monogenic diseases can be largely traced to intragenic variation; however, recent advances in clinical detection and gene sequencing have uncovered the emerging role of non-allelic variation (i.e. genetic trans-modifiers) in shaping disease phenotypes. Identifying these associations are not only of significant diagnostic value, but also provides scientific insight into the expanded molecular etiology of rare diseases. This reports describes the discordant clinical manifestation of a family segregating mutations in ABCA4 and PROM1. Methods: Three patients across a two generation family underwent multimodal imaging and functional testing of the retina including color photography, fundus autofluorescence (AF), spectral domain-optical coherence tomography (SD-OCT) and full-field electroretinography (ffERG). Genetic characterization was carried out by direct Sanger and whole exome sequencing. Results: Clinical examination revealed similar retinal degenerative phenotypes in the proband and her mother. Despite being younger, the proband's phenotype was more advanced and exhibited additional features related to Stargardt disease not found in the mother. Whole exome sequencing identified a pathogenic missense variant in PROM1, c.400C > T, p.(Arg134Cys), as the underlying cause of retinal disease in both the proband and mother. Sequencing of the ABCA4 locus uncovered a single disease-causing variant, c.5714 + 5G > A in the daughter segregating from the father who, surprisingly, also exhibited very subtle disease changes associated with STGD1 despite being a heterozygous carrier. Conclusions: Harboring an additional heterozygous ABCA4 mutation increases severity and confers STGD1-like features in patients with PROM1 disease which provides supporting evidence for their shared pathophysiology and potential treatment prospects.
Keywords: ABCA4; PROM1; cone-rod dystrophy; family; modifier.
Conflict of interest statement
Figures
References
-
- McCulloch DL, Marmor MF, Brigell MG, Hamilton R, Holder GE, Tzekov R, et al. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc Ophthalmol. 2015;130(1):1–12. - PubMed
-
- Jagadeesh KA, Wenger AM, Berger MJ, Guturu H, Stenson PD, Cooper DN, et al. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet. 2016;48(12):1581–6. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials