

Dilated cardiomyopathy mutation in the converter domain of human cardiac myosin alters motor activity and response to omecamtiv mecarbil
- PMID: 31578282
- PMCID: PMC6873177
- DOI: 10.1074/jbc.RA119.010217
Dilated cardiomyopathy mutation in the converter domain of human cardiac myosin alters motor activity and response to omecamtiv mecarbil
Abstract
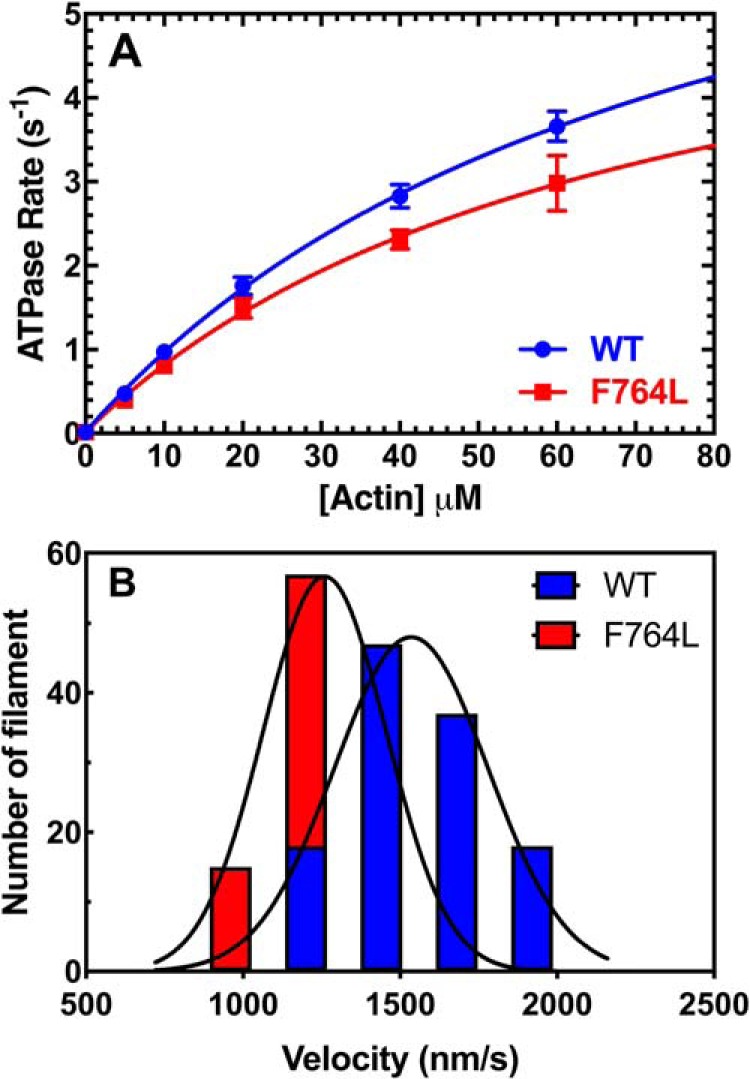
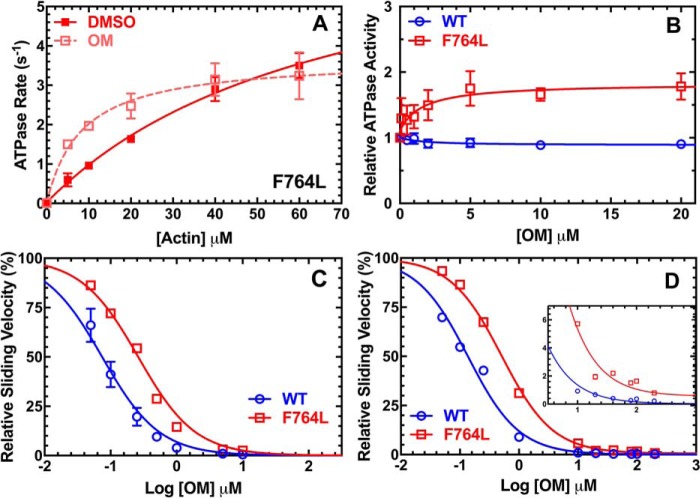
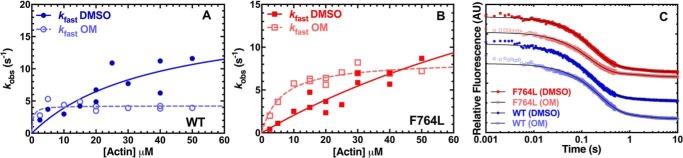
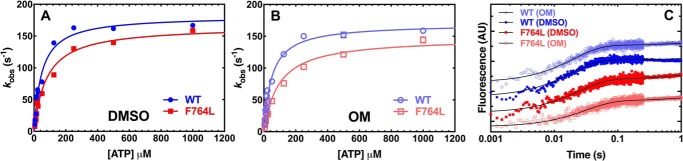
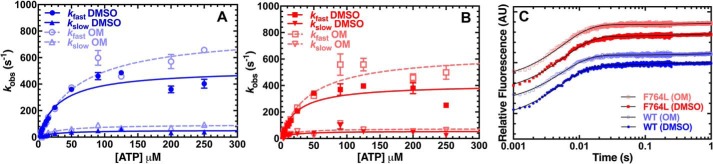
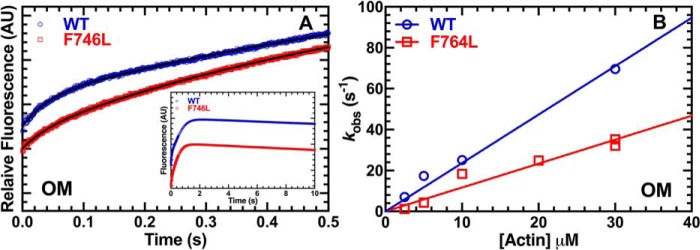
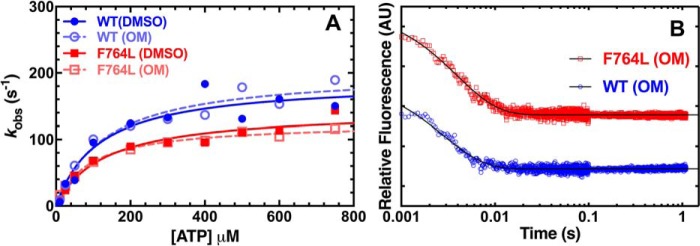
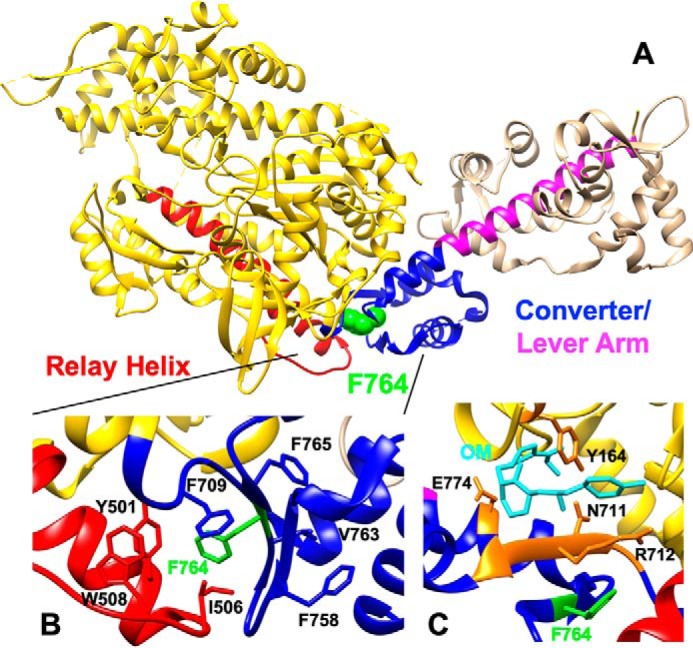
We investigated a dilated cardiomyopathy (DCM) mutation (F764L) in human β-cardiac myosin by determining its motor properties in the presence and absence of the heart failure drug omecamtive mecarbil (OM). The mutation is located in the converter domain, a key region of communication between the catalytic motor and lever arm in myosins, and is nearby but not directly in the OM-binding site. We expressed and purified human β-cardiac myosin subfragment 1 (M2β-S1) containing the F764L mutation, and compared it to WT with in vitro motility as well as steady-state and transient kinetics measurements. In the absence of OM we demonstrate that the F764L mutation does not significantly change maximum actin-activated ATPase activity but slows actin sliding velocity (15%) and the actomyosin ADP release rate constant (25%). The transient kinetic analysis without OM demonstrates that F764L has a similar duty ratio as WT in unloaded conditions. OM is known to enhance force generation in cardiac muscle while it inhibits the myosin power stroke and enhances actin-attachment duration. We found that OM has a reduced impact on F764L ATPase and sliding velocity compared with WT. Specifically, the EC50 for OM induced inhibition of in vitro motility was 3-fold weaker in F764L. Also, OM reduces maximum actin-activated ATPase 2-fold in F764L, compared with 4-fold with WT. Overall, our results suggest that F764L attenuates the impact of OM on actin-attachment duration and/or the power stroke. Our work highlights the importance of mutation-specific considerations when pursuing small molecule therapies for cardiomyopathies.
Keywords: actin; cardiomyopathy; enzyme kinetics; heart failure; myosin; omecamtiv mecarbil.
© 2019 Tang et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures

References
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
