

KIAA0317 regulates pulmonary inflammation through SOCS2 degradation
- PMID: 31578312
- PMCID: PMC6795399
- DOI: 10.1172/jci.insight.129110
KIAA0317 regulates pulmonary inflammation through SOCS2 degradation
Abstract
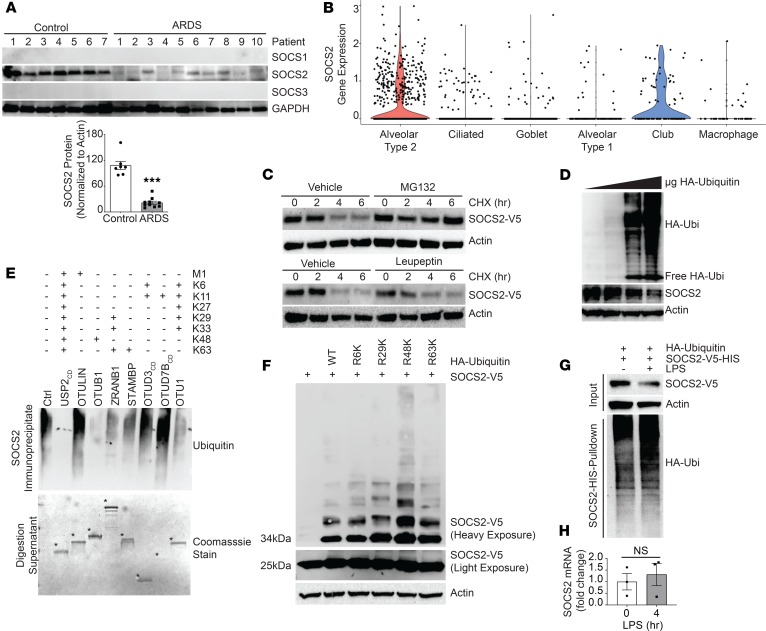
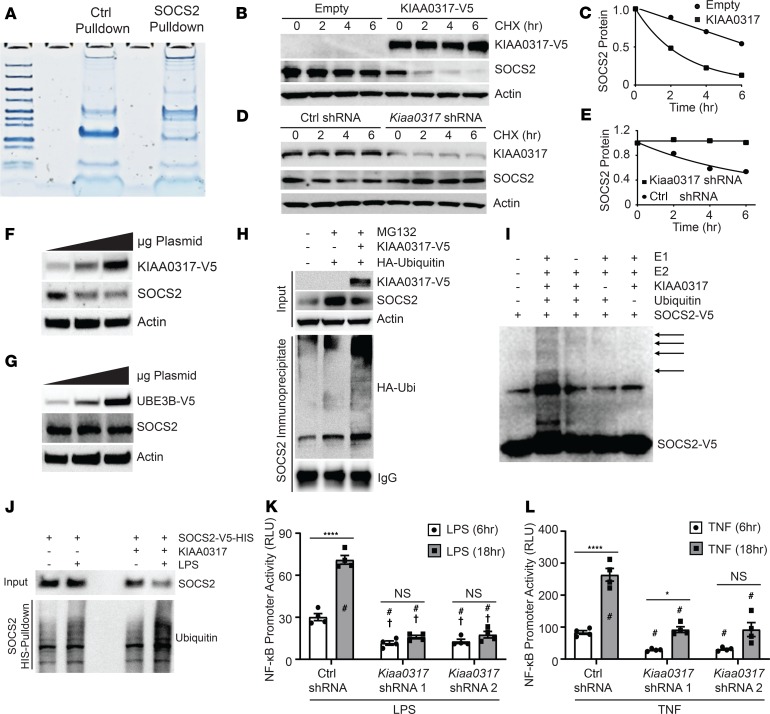
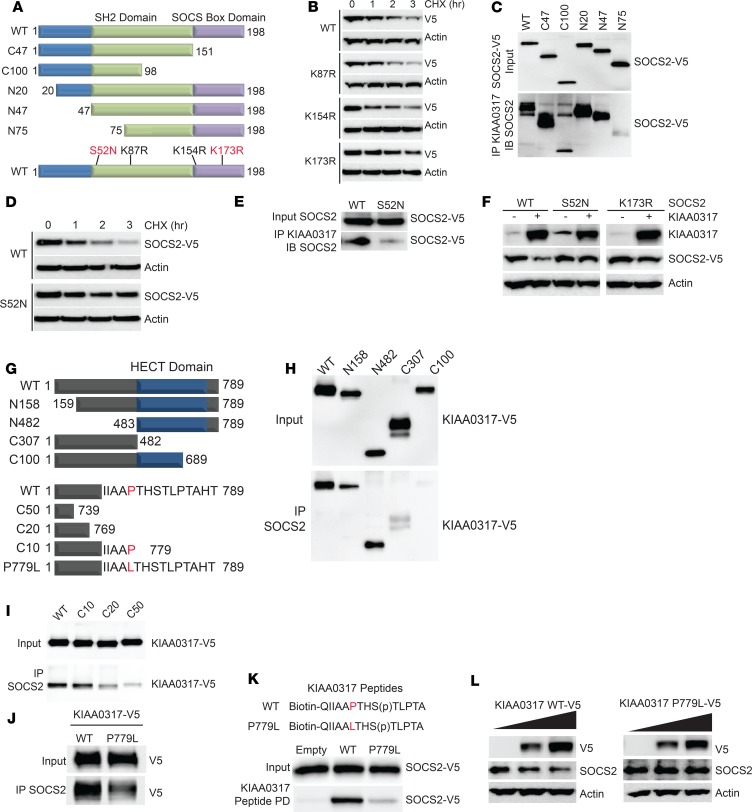
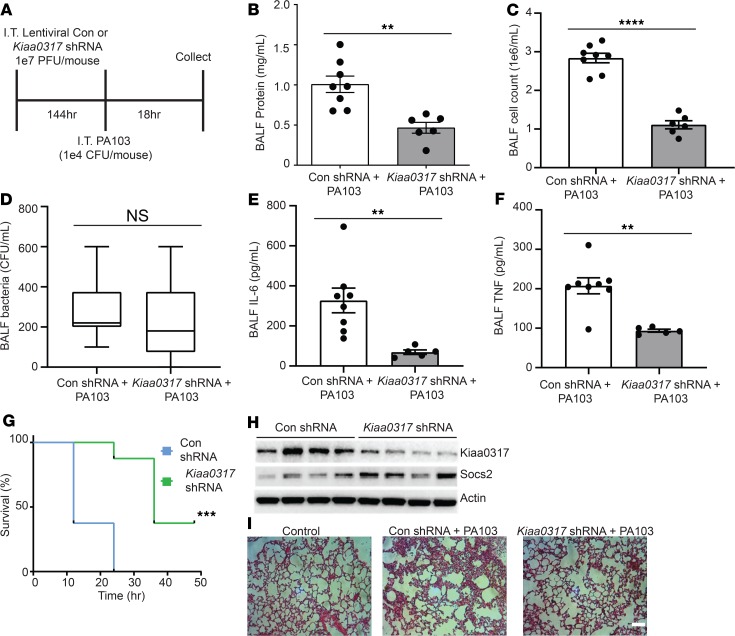
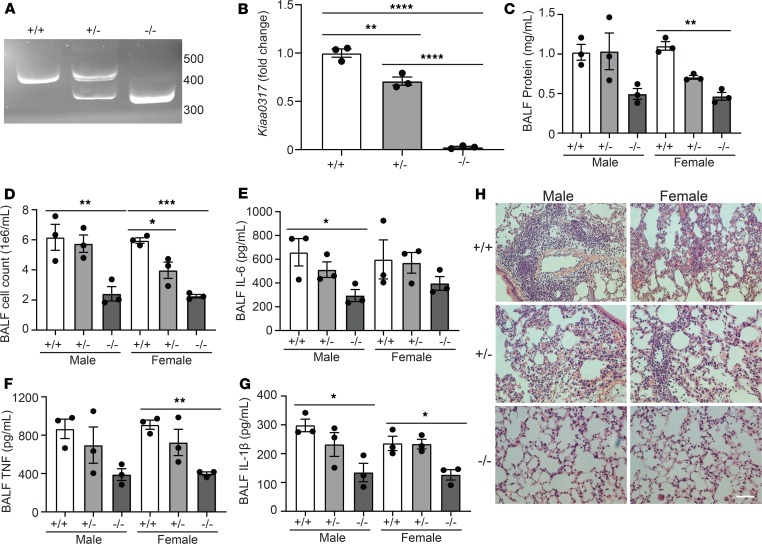
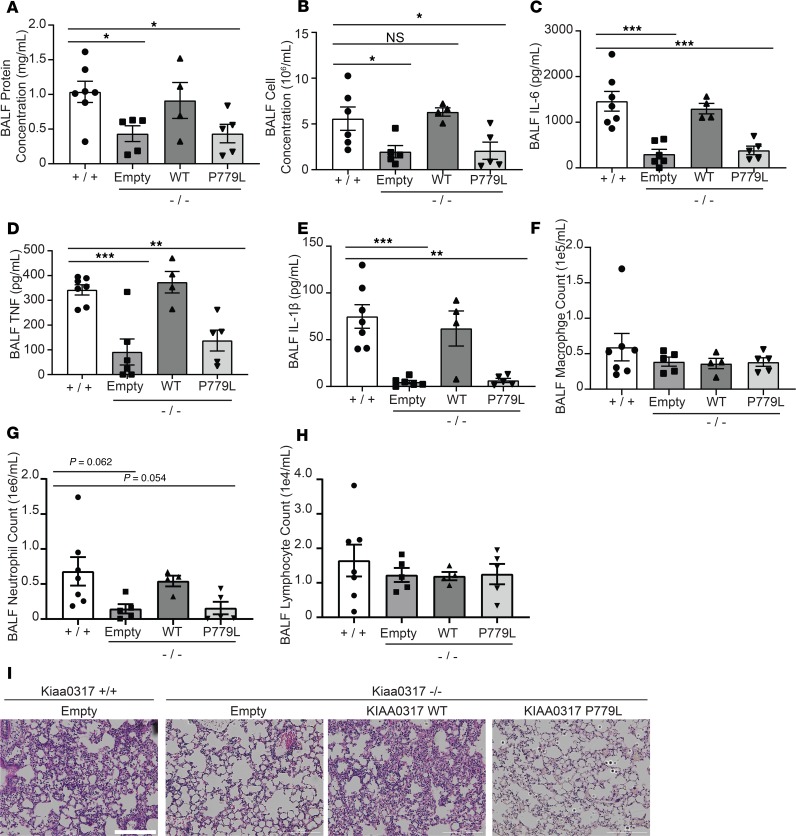
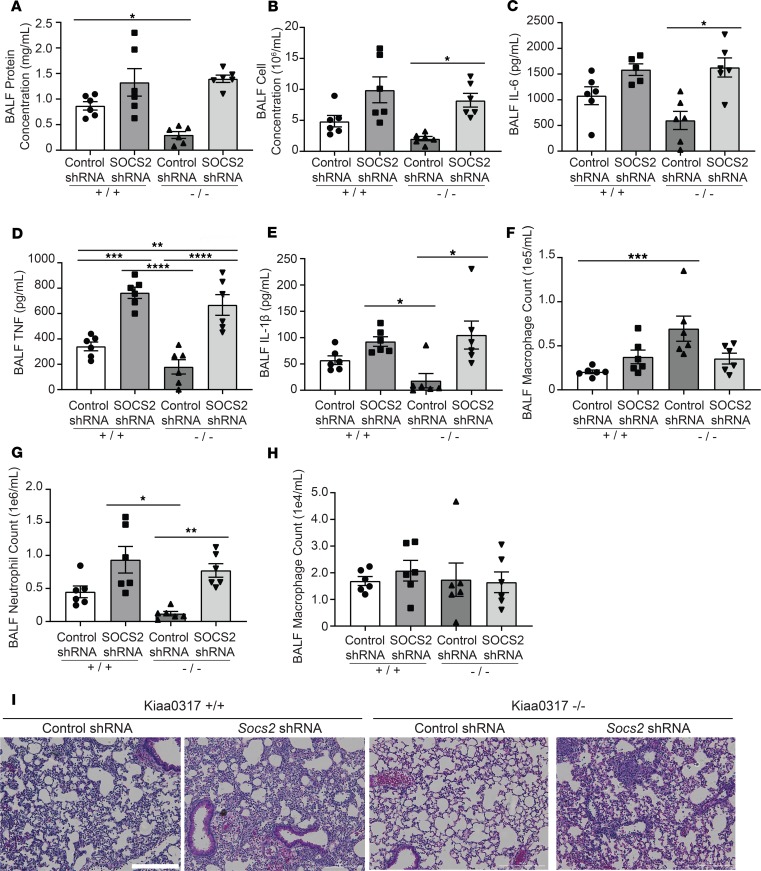
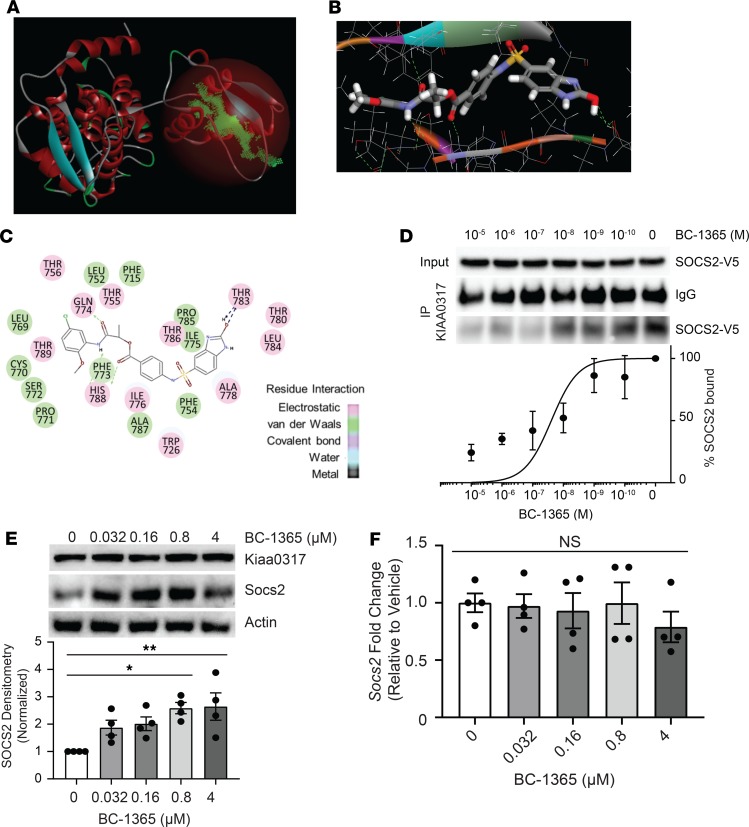
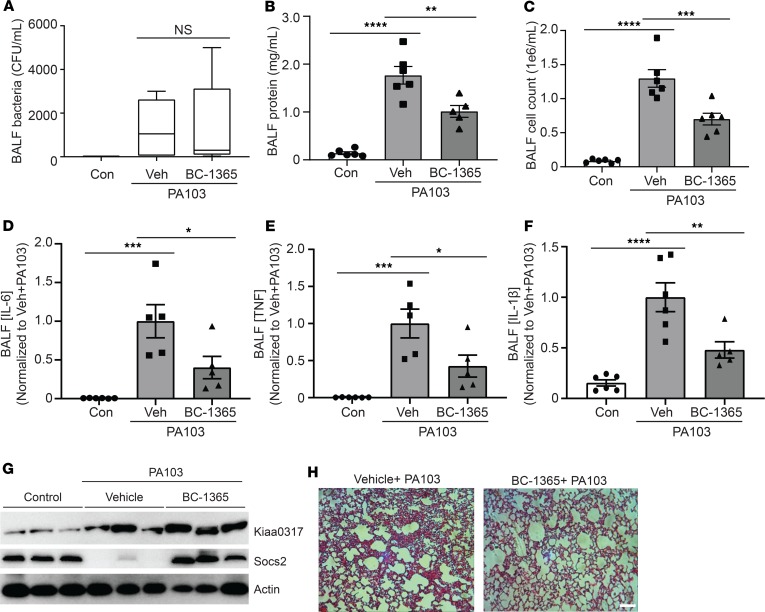
Dysregulated proinflammatory cytokine release has been implicated in the pathogenesis of several life-threatening acute lung illnesses such as pneumonia, sepsis, and acute respiratory distress syndrome. Suppressors of cytokine signaling proteins, particularly SOCS2, have recently been described as antiinflammatory mediators. However, the regulation of SOCS2 protein has not been described. Here we describe a mechanism of SOCS2 regulation by the action of the ubiquitin E3 ligase KIAA0317. KIAA0317-mediated degradation of SOCS2 exacerbated inflammation in vitro, and depletion of KIAA0317 in vivo ameliorated pulmonary inflammation. KIAA0317-knockout mice exhibited resistance to LPS-induced pulmonary inflammation, while KIAA03017 reexpression mitigated this effect. We uncovered a small molecule inhibitor of KIAA0317 protein (BC-1365) that prevented SOCS2 degradation and attenuated LPS- and P. aeruginosa-induced lung inflammation in vivo. These studies show KIAA0317 to be a critical mediator of pulmonary inflammation through its degradation of SOCS2 and a potential candidate target for therapeutic inhibition.
Keywords: Inflammation; Innate immunity; Pulmonology; Ubiquitin-proteosome system.
Conflict of interest statement
Figures
Similar articles
-
A growth hormone receptor SNP promotes lung cancer by impairment of SOCS2-mediated degradation.Oncogene. 2018 Jan 25;37(4):489-501. doi: 10.1038/onc.2017.352. Epub 2017 Oct 2. Oncogene. 2018. PMID: 28967904 Free PMC article.
-
Leveraging Therapeutic Proteins and Peptides from Lumbricus Earthworms: Targeting SOCS2 E3 Ligase for Cardiovascular Therapy through Molecular Dynamics Simulations.Int J Mol Sci. 2024 Oct 8;25(19):10818. doi: 10.3390/ijms251910818. Int J Mol Sci. 2024. PMID: 39409145 Free PMC article.
-
KIAA0317 regulates SOCS1 stability to ameliorate colonic inflammation.FEBS J. 2023 Aug;290(15):3802-3811. doi: 10.1111/febs.16780. Epub 2023 Apr 7. FEBS J. 2023. PMID: 36938956 Free PMC article.
-
Parkin regulates lipopolysaccharide-induced proinflammatory responses in acute lung injury.Transl Res. 2017 Mar;181:71-82. doi: 10.1016/j.trsl.2016.09.002. Epub 2016 Sep 13. Transl Res. 2017. PMID: 27693468
-
The many faces of the SOCS box.Cytokine Growth Factor Rev. 2008 Oct-Dec;19(5-6):371-81. doi: 10.1016/j.cytogfr.2008.08.006. Epub 2008 Oct 22. Cytokine Growth Factor Rev. 2008. PMID: 18948053 Review.
Cited by
-
An IL-17F.S65L Knock-In Mouse Reveals Similarities and Differences in IL-17F Function in Oral Candidiasis: A New Tool to Understand IL-17F.J Immunol. 2020 Aug 1;205(3):720-730. doi: 10.4049/jimmunol.2000394. Epub 2020 Jun 29. J Immunol. 2020. PMID: 32601099 Free PMC article.
-
Epigenetics as a mediator of genetic risk in osteoarthritis: role during development, homeostasis, aging, and disease progression.Am J Physiol Cell Physiol. 2023 May 1;324(5):C1078-C1088. doi: 10.1152/ajpcell.00574.2022. Epub 2023 Mar 27. Am J Physiol Cell Physiol. 2023. PMID: 36971423 Free PMC article. Review.
-
Redefining the catalytic HECT domain boundaries for the HECT E3 ubiquitin ligase family.Biosci Rep. 2022 Oct 28;42(10):BSR20221036. doi: 10.1042/BSR20221036. Biosci Rep. 2022. PMID: 36111624 Free PMC article.
-
The Emerging Roles of Tripartite Motif Proteins (TRIMs) in Acute Lung Injury.J Immunol Res. 2021 Oct 19;2021:1007126. doi: 10.1155/2021/1007126. eCollection 2021. J Immunol Res. 2021. PMID: 34712740 Free PMC article. Review.
-
A genomic perspective of the aging human and mouse lung with a focus on immune response and cellular senescence.Immun Ageing. 2023 Nov 6;20(1):58. doi: 10.1186/s12979-023-00373-5. Immun Ageing. 2023. PMID: 37932771 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
