

Transcriptional and epigenetic regulation of macrophages in atherosclerosis
- PMID: 31578516
- PMCID: PMC7770754
- DOI: 10.1038/s41569-019-0265-3
Transcriptional and epigenetic regulation of macrophages in atherosclerosis
Abstract
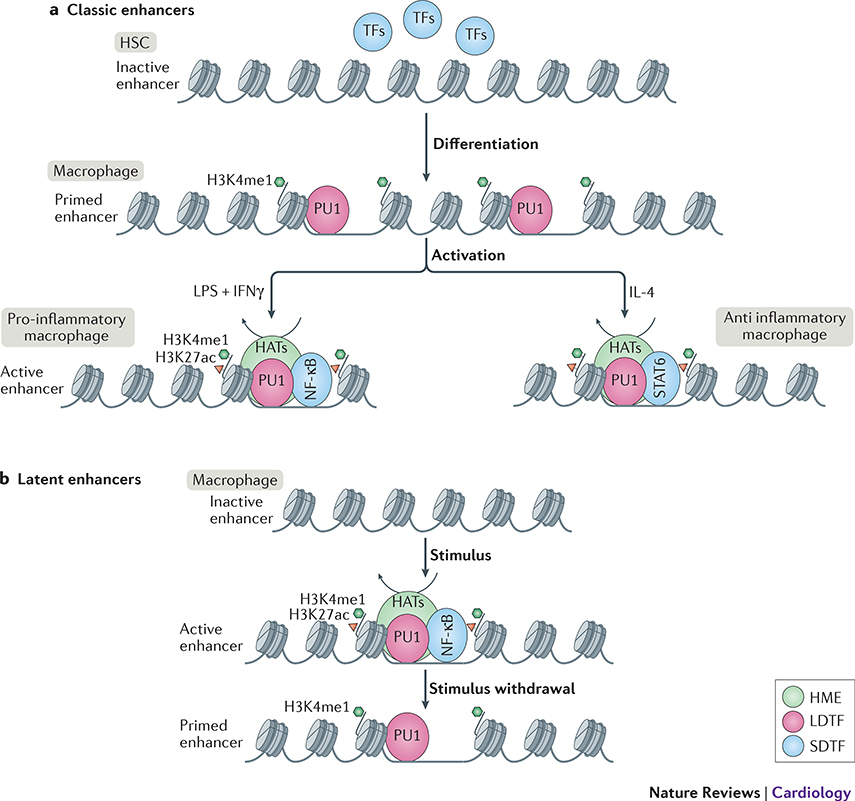
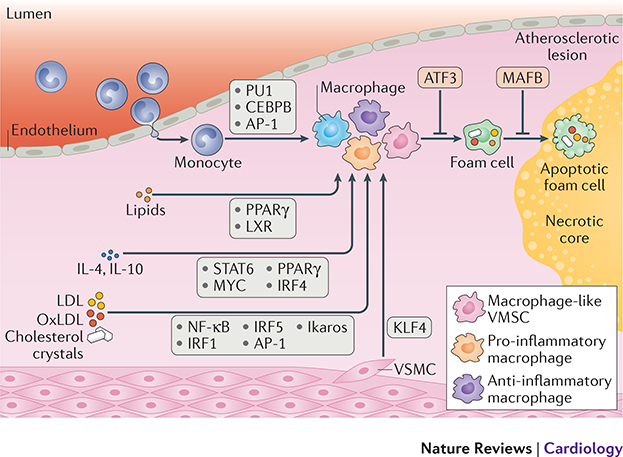
Monocytes and macrophages provide defence against pathogens and danger signals. These cells respond to stimulation in a fast and stimulus-specific manner by utilizing complex cascaded activation by lineage-determining and signal-dependent transcription factors. The complexity of the functional response is determined by interactions between triggered transcription factors and depends on the microenvironment and interdependent signalling cascades. Dysregulation of macrophage phenotypes is a major driver of various diseases such as atherosclerosis, rheumatoid arthritis and type 2 diabetes mellitus. Furthermore, exposure of monocytes, which are macrophage precursor cells, to certain stimuli can lead to a hypo-inflammatory tolerized phenotype or a hyper-inflammatory trained phenotype in a macrophage. In atherosclerosis, macrophages and monocytes are exposed to inflammatory cytokines, oxidized lipids, cholesterol crystals and other factors. All these stimuli induce not only a specific transcriptional response but also interact extensively, leading to transcriptional and epigenetic heterogeneity of macrophages in atherosclerotic plaques. Targeting the epigenetic landscape of plaque macrophages can be a powerful therapeutic tool to modulate pro-atherogenic phenotypes and reduce the rate of plaque formation. In this Review, we discuss the emerging role of transcription factors and epigenetic remodelling in macrophages in the context of atherosclerosis and inflammation, and provide a comprehensive overview of epigenetic enzymes and transcription factors that are involved in macrophage activation.
Conflict of interest statement
Competing interests
None
Figures

References
-
- Ginhoux F, Schultze JL, Murray PJ, Ochando J, Biswas SK. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat Immunol. Nature Publishing Group; 2016. January;17(1):34–40. - PubMed
-
- Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, De Nardo D, Gohel TD, Emde M, Schmidleithner L, Ganesan H, Nino-Castro A, Mallmann MR, Labzin L, Theis H, Kraut M, Beyer M, Latz E, Freeman TC, Ulas T, Schultze JL. Transcriptome-Based Network Analysis Reveals a Spectrum Model of Human Macrophage Activation. Immunity. Elsevier Inc; 2014. February 20;40(2):274–88. - PMC - PubMed
-
- Colin S, Chinetti-Gbaguidi G, Staels B. Macrophage phenotypes in atherosclerosis. Immunol Rev. John Wiley & Sons, Ltd (10.1111); 2014. November;262(1):153–66. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
