

Binding-induced folding under unfolding conditions: Switching between induced fit and conformational selection mechanisms
- PMID: 31582563
- PMCID: PMC6851327
- DOI: 10.1074/jbc.RA119.009742
Binding-induced folding under unfolding conditions: Switching between induced fit and conformational selection mechanisms
Abstract
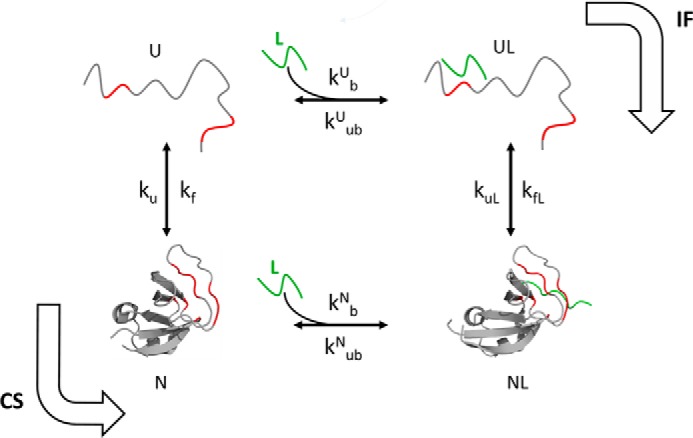
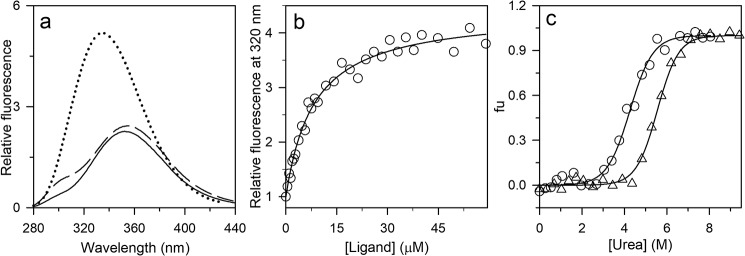
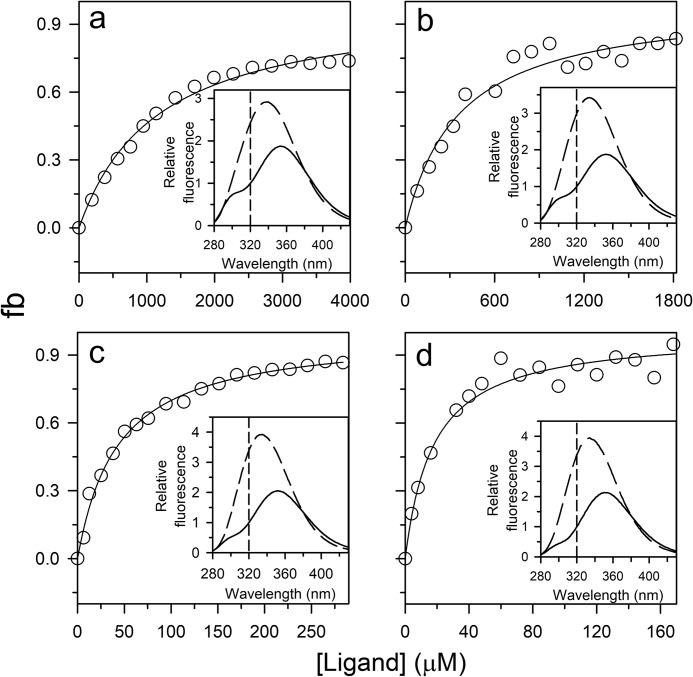
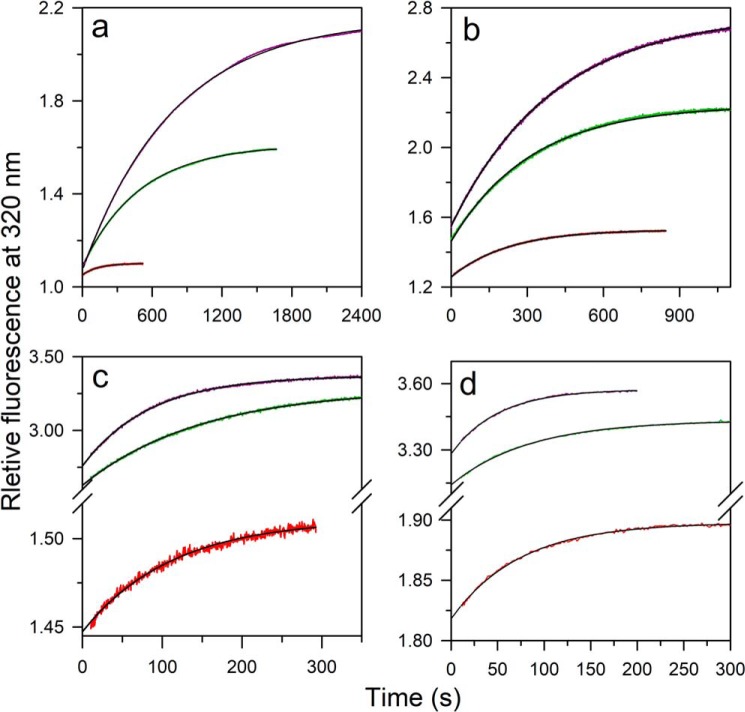
The chemistry of protein-ligand binding is the basis of virtually every biological process. Ligand binding can be essential for a protein to function in the cell by stabilizing or altering the conformation of a protein, particularly for partially or completely unstructured proteins. However, the mechanisms by which ligand binding impacts disordered proteins or influences the role of disorder in protein folding is not clear. To gain insight into this question, the mechanism of folding induced by the binding of a Pro-rich peptide ligand to the SH3 domain of phosphatidylinositol 3-kinase unfolded in the presence of urea has been studied using kinetic methods. Under strongly denaturing conditions, folding was found to follow a conformational selection (CS) mechanism. However, under mildly denaturing conditions, a ligand concentration-dependent switch in the mechanism was observed. The folding mechanism switched from being predominantly a CS mechanism at low ligand concentrations to being predominantly an induced fit (IF) mechanism at high ligand concentrations. The switch in the mechanism manifests itself as an increase in the reaction flux along the IF pathway at high ligand concentrations. The results indicate that, in the case of intrinsically disordered proteins too, the folding mechanism is determined by the concentration of the ligand that induces structure formation.
Keywords: Src homology 3 domain (SH3 domain); conformational selection; induced fit; intrinsically disordered protein; ligand-binding protein; peptides; protein folding; reaction flux.
© 2019 Sen and Udgaonkar.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
References
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Miscellaneous
