

Highly structured homolog pairing reflects functional organization of the Drosophila genome
- PMID: 31582763
- PMCID: PMC6776532
- DOI: 10.1038/s41467-019-12208-3
Highly structured homolog pairing reflects functional organization of the Drosophila genome
Abstract
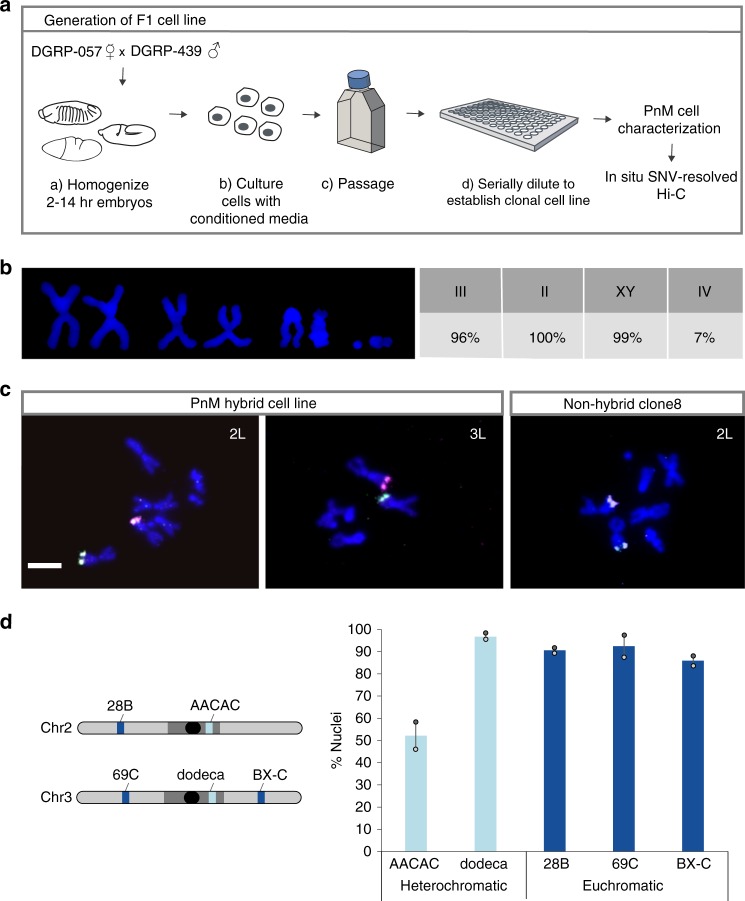
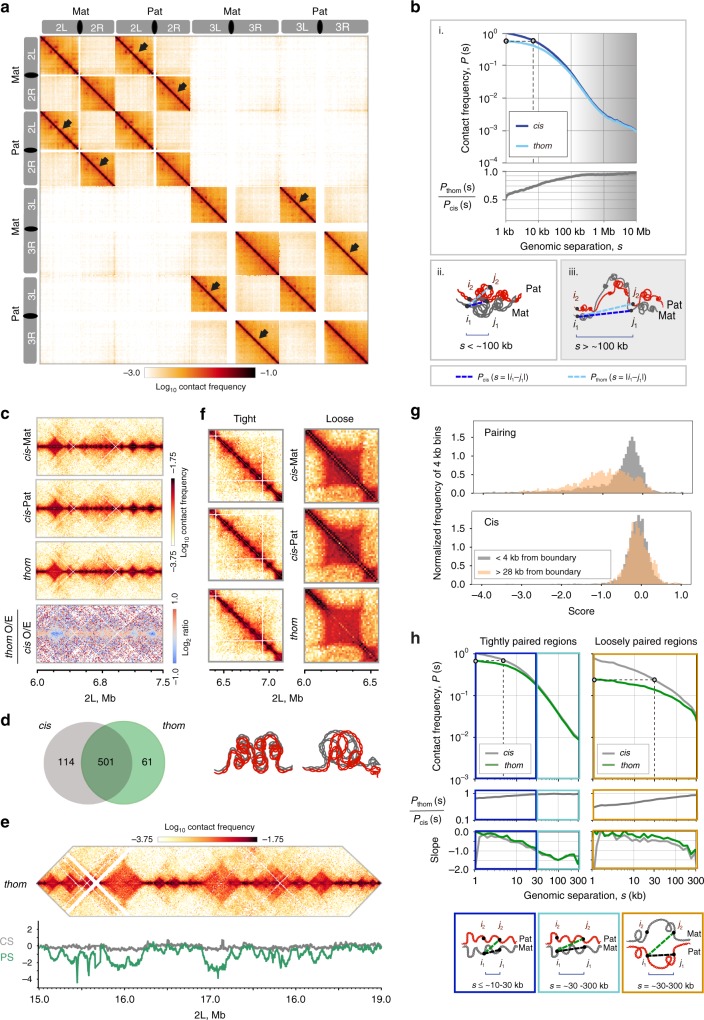
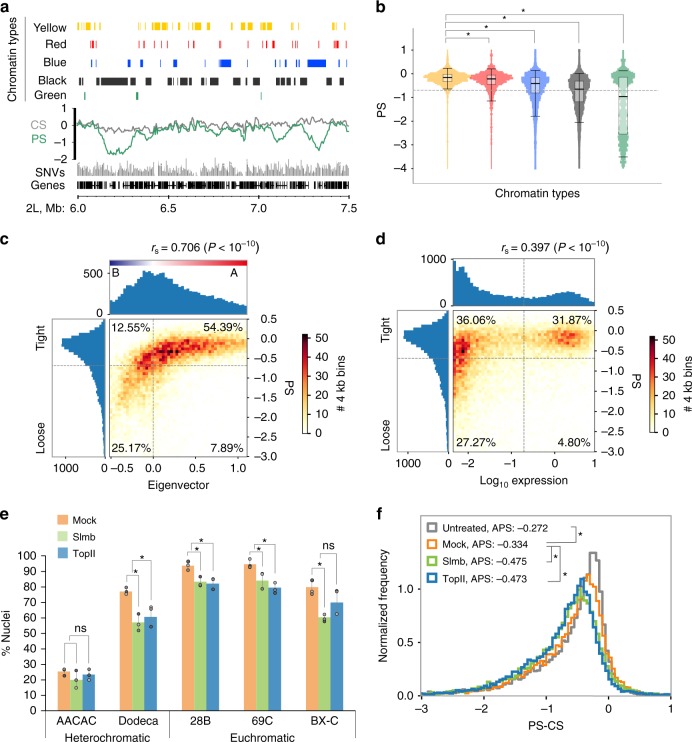
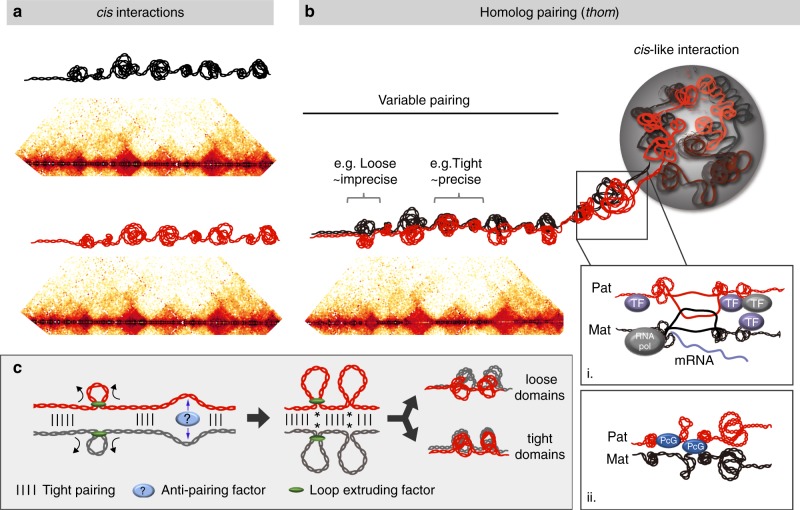
Trans-homolog interactions have been studied extensively in Drosophila, where homologs are paired in somatic cells and transvection is prevalent. Nevertheless, the detailed structure of pairing and its functional impact have not been thoroughly investigated. Accordingly, we generated a diploid cell line from divergent parents and applied haplotype-resolved Hi-C, showing that homologs pair with varying precision genome-wide, in addition to establishing trans-homolog domains and compartments. We also elucidate the structure of pairing with unprecedented detail, observing significant variation across the genome and revealing at least two forms of pairing: tight pairing, spanning contiguous small domains, and loose pairing, consisting of single larger domains. Strikingly, active genomic regions (A-type compartments, active chromatin, expressed genes) correlated with tight pairing, suggesting that pairing has a functional implication genome-wide. Finally, using RNAi and haplotype-resolved Hi-C, we show that disruption of pairing-promoting factors results in global changes in pairing, including the disruption of some interaction peaks.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
