

Engineering Phage Host-Range and Suppressing Bacterial Resistance through Phage Tail Fiber Mutagenesis
- PMID: 31585083
- PMCID: PMC6924272
- DOI: 10.1016/j.cell.2019.09.015
Engineering Phage Host-Range and Suppressing Bacterial Resistance through Phage Tail Fiber Mutagenesis
Abstract
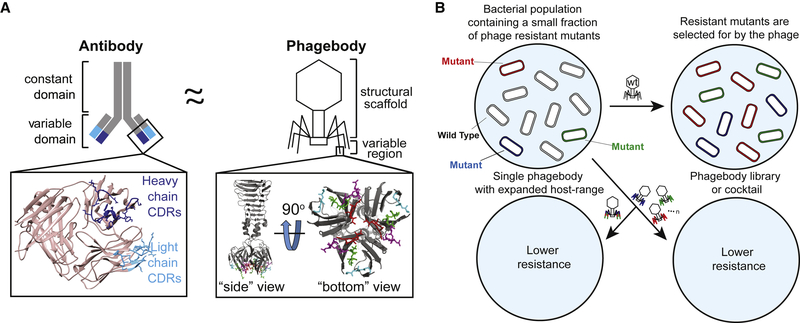
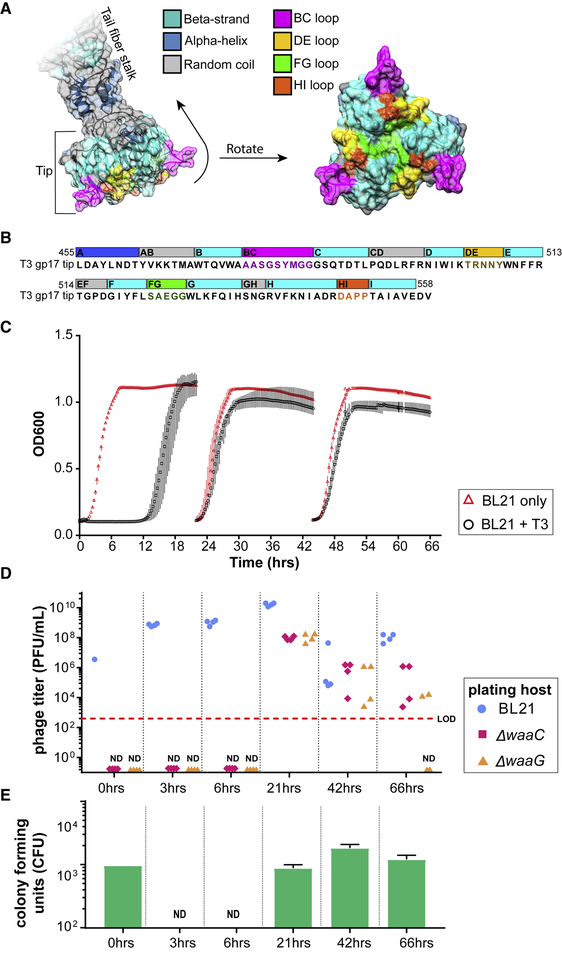
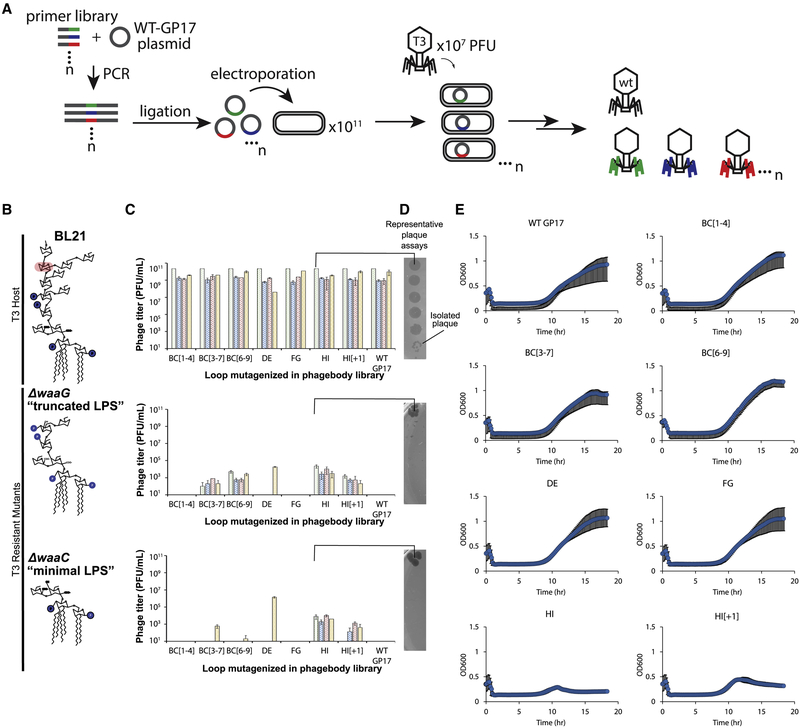
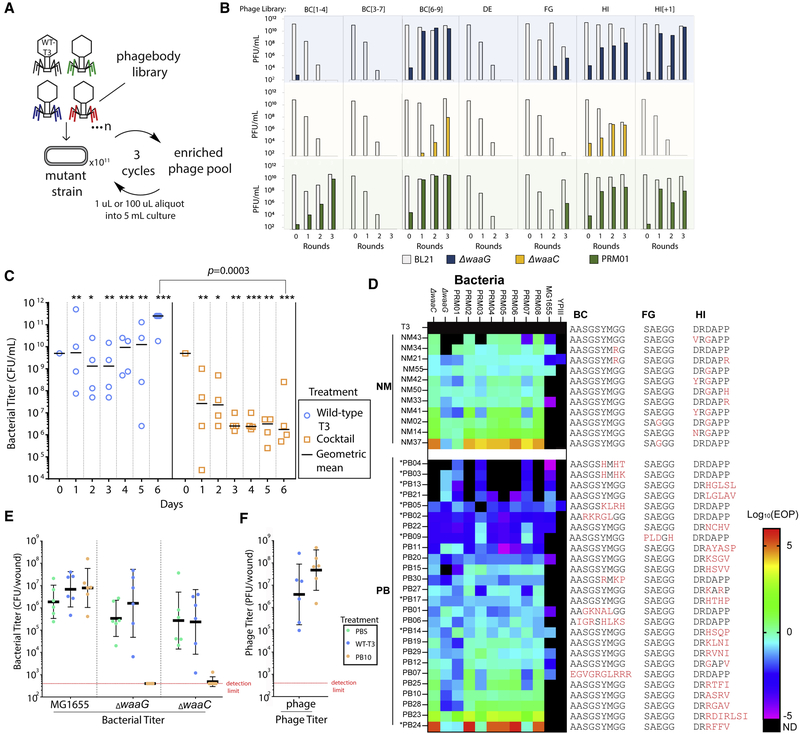
The rapid emergence of antibiotic-resistant infections is prompting increased interest in phage-based antimicrobials. However, acquisition of resistance by bacteria is a major issue in the successful development of phage therapies. Through natural evolution and structural modeling, we identified host-range-determining regions (HRDRs) in the T3 phage tail fiber protein and developed a high-throughput strategy to genetically engineer these regions through site-directed mutagenesis. Inspired by antibody specificity engineering, this approach generates deep functional diversity while minimizing disruptions to the overall tail fiber structure, resulting in synthetic "phagebodies." We showed that mutating HRDRs yields phagebodies with altered host-ranges, and select phagebodies enable long-term suppression of bacterial growth in vitro, by preventing resistance appearance, and are functional in vivo using a murine model. We anticipate that this approach may facilitate the creation of next-generation antimicrobials that slow resistance development and could be extended to other viral scaffolds for a broad range of applications.
Keywords: antibody; antimicrobial; bacteriophage; evolution; host-range; phage; resistance; synthetic biology; tail fiber; virus.
Copyright © 2019 Elsevier Inc. All rights reserved.
Conflict of interest statement
DECLARATION OF INTERESTS
T.K.L. is a co-founder of Senti Biosciences, Synlogic, Engine Biosciences, Tango Therapeutics, Corvium, BiomX, and Eligo Biosciences. T.K.L. also holds financial interests in nest.bio, Ampliphi, IndieBio, MedicusTek, Quark Biosciences, and Personal Genomics. T.K.L., K.Y., S.L., and H.A. have patents related to this work.
Figures
Comment in
-
Learning from Antibodies: Phage Host-Range Engineering.Cell Host Microbe. 2019 Oct 9;26(4):445-446. doi: 10.1016/j.chom.2019.09.010. Cell Host Microbe. 2019. PMID: 31600495
References
-
- Arnold K, Bordoli L, Kopp J, and Schwede T (2006). The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201. - PubMed
-
- Beck A, Wurch T, Bailly C, and Corvaia N (2010). Strategies and challenges for the next generation of therapeutic antibodies. Nat. Rev. Immunol 10, 345–352. - PubMed
-
- Bertozzi Silva J, Storms Z, and Sauvageau D (2016). Host receptors for bacteriophage adsorption. FEMS Microbiol. Lett 363, fnw002. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
