

Metagenomic Signatures of Gut Infections Caused by Different Escherichia coli Pathotypes
- PMID: 31585992
- PMCID: PMC6881795
- DOI: 10.1128/AEM.01820-19
Metagenomic Signatures of Gut Infections Caused by Different Escherichia coli Pathotypes
Abstract
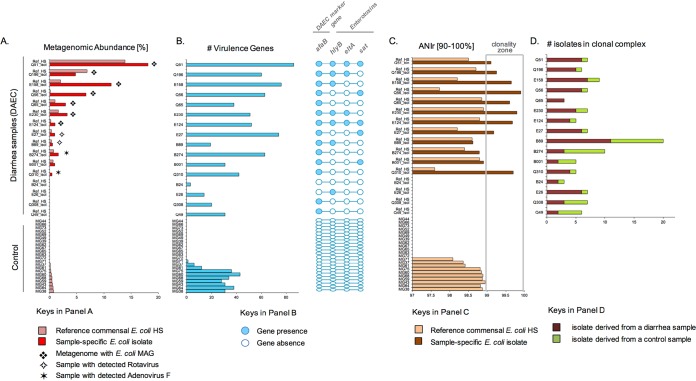
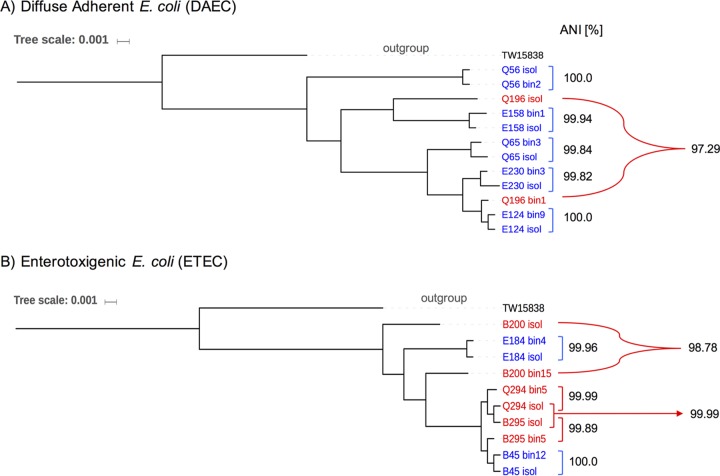
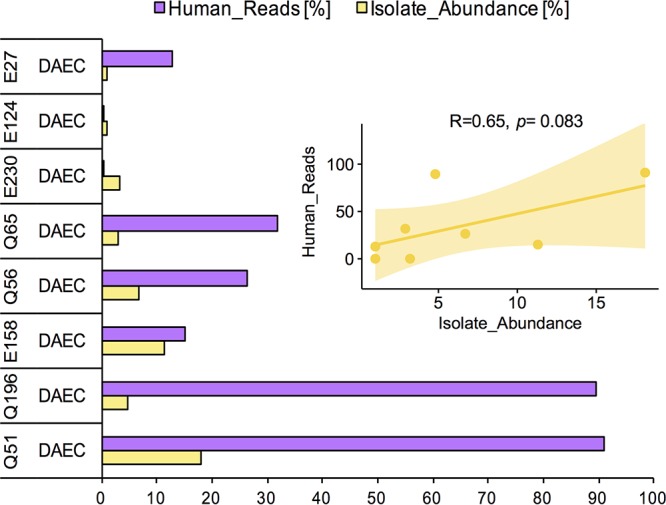
Escherichia coli is a leading contributor to infectious diarrhea and child mortality worldwide, but it remains unknown how alterations in the gut microbiome vary for distinct E. coli pathotype infections and whether these signatures can be used for diagnostic purposes. Further, the majority of enteric diarrheal infections are not diagnosed with respect to their etiological agent(s) due to technical challenges. To address these issues, we devised a novel approach that combined traditional, isolate-based and molecular-biology techniques with metagenomics analysis of stool samples and epidemiological data. Application of this pipeline to children enrolled in a case-control study of diarrhea in Ecuador showed that, in about half of the cases where an E. coli pathotype was detected by culture and PCR, E. coli was likely not the causative agent based on the metagenome-derived low relative abundance, the level of clonality, and/or the virulence gene content. Our results also showed that diffuse adherent E. coli (DAEC), a pathotype that is generally underrepresented in previous studies of diarrhea and thus, thought not to be highly virulent, caused several small-scale diarrheal outbreaks across a rural to urban gradient in Ecuador. DAEC infections were uniquely accompanied by coelution of large amounts of human DNA and conferred significant shifts in the gut microbiome composition relative to controls or infections caused by other E. coli pathotypes. Our study shows that diarrheal infections can be efficiently diagnosed for their etiological agent and categorized based on their effects on the gut microbiome using metagenomic tools, which opens new possibilities for diagnostics and treatment.IMPORTANCEE. coli infectious diarrhea is an important contributor to child mortality worldwide. However, diagnosing and thus treating E. coli infections remain challenging due to technical and other reasons associated with the limitations of the traditional culture-based techniques and the requirement to apply Koch's postulates. In this study, we integrated traditional microbiology techniques with metagenomics and epidemiological data in order to identify cases of diarrhea where E. coli was most likely the causative disease agent and evaluate specific signatures in the disease-state gut microbiome that distinguish between diffuse adherent, enterotoxigenic, and enteropathogenic E. coli pathotypes. Therefore, our methodology and results should be highly relevant for diagnosing and treating diarrheal infections and have important applications in public health.
Keywords: 16S rRNA; Ecuador; Escherichia coli; clinical metagenomics; gut microbiome; infectious diarrhea; metagenomics; pathotypes.
Copyright © 2019 American Society for Microbiology.
Figures


Similar articles
-
Comparison of metagenomic and traditional methods for diagnosis of E. coli enteric infections.mBio. 2024 Apr 10;15(4):e0342223. doi: 10.1128/mbio.03422-23. Epub 2024 Mar 15. mBio. 2024. PMID: 38488359 Free PMC article.
-
Distribution of Escherichia coli Pathotypes along an Urban-Rural Gradient in Ecuador.Am J Trop Med Hyg. 2023 Aug 7;109(3):559-567. doi: 10.4269/ajtmh.23-0167. Print 2023 Sep 6. Am J Trop Med Hyg. 2023. PMID: 37549901 Free PMC article.
-
Why are so many enteric pathogen infections asymptomatic? Pathogen and gut microbiome characteristics associated with diarrhea symptoms and carriage of diarrheagenic E. coli in northern Ecuador.Gut Microbes. 2023 Dec;15(2):2281010. doi: 10.1080/19490976.2023.2281010. Epub 2023 Nov 22. Gut Microbes. 2023. PMID: 37992406 Free PMC article.
-
Updates on defining and detecting diarrheagenic Escherichia coli pathotypes.Curr Opin Infect Dis. 2020 Oct;33(5):372-380. doi: 10.1097/QCO.0000000000000665. Curr Opin Infect Dis. 2020. PMID: 32773499 Free PMC article. Review.
-
[Virulence mechanisms of enteropathogenic Escherichia coli].Rev Chilena Infectol. 2016 Aug;33(4):438-450. doi: 10.4067/S0716-10182016000400009. Rev Chilena Infectol. 2016. PMID: 27905628 Review. Spanish.
Cited by
-
Homologous Escherichia coli Identified in Cerebrospinal Fluid and Bloodstream.Front Cell Infect Microbiol. 2021 Sep 10;11:674235. doi: 10.3389/fcimb.2021.674235. eCollection 2021. Front Cell Infect Microbiol. 2021. PMID: 34568083 Free PMC article.
-
Strain-level epidemiology of microbial communities and the human microbiome.Genome Med. 2020 Aug 13;12(1):71. doi: 10.1186/s13073-020-00765-y. Genome Med. 2020. PMID: 32791981 Free PMC article. Review.
-
Gut microbiome, enteric infections and child growth across a rural-urban gradient: protocol for the ECoMiD prospective cohort study.BMJ Open. 2021 Oct 22;11(10):e046241. doi: 10.1136/bmjopen-2020-046241. BMJ Open. 2021. PMID: 34686548 Free PMC article.
-
Measuring Environmental Exposure to Enteric Pathogens in Low-Income Settings: Review and Recommendations of an Interdisciplinary Working Group.Environ Sci Technol. 2020 Oct 6;54(19):11673-11691. doi: 10.1021/acs.est.0c02421. Epub 2020 Sep 9. Environ Sci Technol. 2020. PMID: 32813503 Free PMC article. Review.
-
The Reliability of Metagenome-Assembled Genomes (MAGs) in Representing Natural Populations: Insights from Comparing MAGs against Isolate Genomes Derived from the Same Fecal Sample.Appl Environ Microbiol. 2021 Feb 26;87(6):e02593-20. doi: 10.1128/AEM.02593-20. Print 2021 Feb 26. Appl Environ Microbiol. 2021. PMID: 33452027 Free PMC article.
References
-
- Troeger C, Forouzanfar M, Rao PC, Khalil I, Brown A, Reiner RC, Fullman N, Thompson RL, Abajobir A, Ahmed M, Alemayohu MA, Alvis-Guzman N, Amare AT, Antonio CA, Asayesh H, Avokpaho E, Awasthi A, Bacha U, Barac A, Betsue BD, Beyene AS, Boneya DJ, Malta DC, Dandona L, Dandona R, Dubey M, Eshrati B, Fitchett JRA, Gebrehiwot TT, Hailu GB, Horino M, Hotez PJ, Jibat T, Jonas JB, Kasaeian A, Kissoon N, Kotloff K, Koyanagi A, Kumar GA, Rai RK, Lal A, El Razek HMA, Mengistie MA, Moe C, Patton G, Platts-Mills JA, Qorbani M, Ram U, Roba HS, Sanabria J, Sartorius B, Sawhney M, Shigematsu M, Sreeramareddy C, Swaminathan S, Tedla BA, Jagiellonian RT-M, Ukwaja K, Werdecker A, Widdowson M-A, Yonemoto N, El Sayed Zaki M, Lim SS, Naghavi M, Vos T, Hay SI, Murray CJL, Mokdad AH. 2017. Estimates of global, regional, and national morbidity, mortality, and aetiologies of diarrhoeal diseases: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Infect Dis 17:909–948. doi: 10.1016/S1473-3099(17)30276-1. - DOI - PMC - PubMed
-
- Selendy JMH. 2011. Water and sanitation-related diseases and the environment: challenges, interventions, and preventive measures. John Wiley & Sons, Inc, New York, NY.
-
- Devleesschauwer B, Haagsma JA, Mangen M-J, Lake RJ, Havelaar AH. 2018. The global burden of foodborne disease, p 107–122. In Food safety economics. Springer, New York, NY.
-
- Kelly D, Khurram NA, Hickman RA, Pei Z. 2018. Quantitative approach in clinical microbiology: a paradigm shift toward culture-free methods, p 599–615. In Advanced techniques in diagnostic microbiology. Springer, New York, NY.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
