

Realizing the potential of full-length transcriptome sequencing
- PMID: 31587638
- PMCID: PMC6792442
- DOI: 10.1098/rstb.2019.0097
Realizing the potential of full-length transcriptome sequencing
Abstract
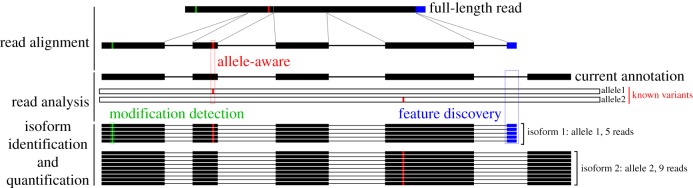
Long-read sequencing holds great potential for transcriptome analysis because it offers researchers an affordable method to annotate the transcriptomes of non-model organisms. This, in turn, will greatly benefit future work on less-researched organisms like unicellular eukaryotes that cannot rely on large consortia to generate these transcriptome annotations. However, to realize this potential, several remaining molecular and computational challenges will have to be overcome. In this review, we have outlined the limitations of short-read sequencing technology and how long-read sequencing technology overcomes these limitations. We have also highlighted the unique challenges still present for long-read sequencing technology and provided some suggestions on how to overcome these challenges going forward. This article is part of a discussion meeting issue 'Single cell ecology'.
Keywords: Oxford Nanopore Technologies; Pacific Biosciences; long-read sequencing; transcriptome analysis.
Conflict of interest statement
Some of the methods we discuss in this review include our own and we have filed patent applications on aspects on them.
Figures



References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources