

Critical assessment of methods of protein structure prediction (CASP)-Round XIII
- PMID: 31589781
- PMCID: PMC6927249
- DOI: 10.1002/prot.25823
Critical assessment of methods of protein structure prediction (CASP)-Round XIII
Abstract
CASP (critical assessment of structure prediction) assesses the state of the art in modeling protein structure from amino acid sequence. The most recent experiment (CASP13 held in 2018) saw dramatic progress in structure modeling without use of structural templates (historically "ab initio" modeling). Progress was driven by the successful application of deep learning techniques to predict inter-residue distances. In turn, these results drove dramatic improvements in three-dimensional structure accuracy: With the proviso that there are an adequate number of sequences known for the protein family, the new methods essentially solve the long-standing problem of predicting the fold topology of monomeric proteins. Further, the number of sequences required in the alignment has fallen substantially. There is also substantial improvement in the accuracy of template-based models. Other areas-model refinement, accuracy estimation, and the structure of protein assemblies-have again yielded interesting results. CASP13 placed increased emphasis on the use of sparse data together with modeling and chemical crosslinking, SAXS, and NMR all yielded more mature results. This paper summarizes the key outcomes of CASP13. The special issue of PROTEINS contains papers describing the CASP13 assessments in each modeling category and contributions from the participants.
Keywords: CASP; community wide experiment; protein structure prediction.
© 2019 Wiley Periodicals, Inc.
Figures
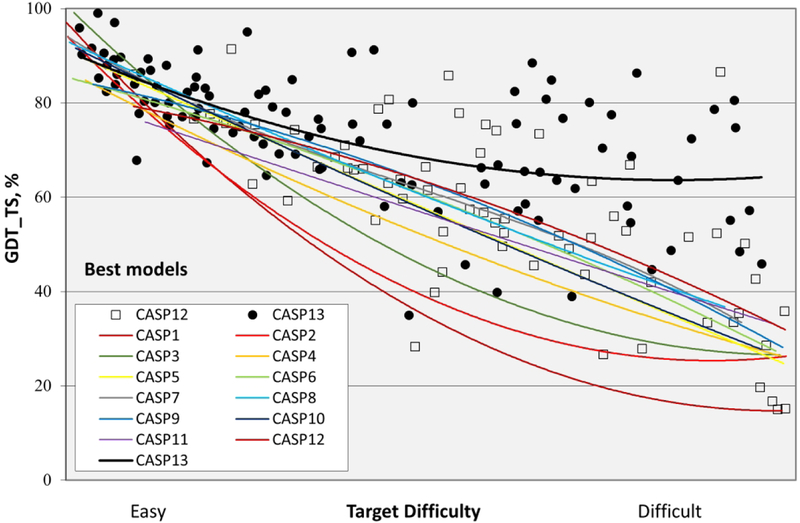
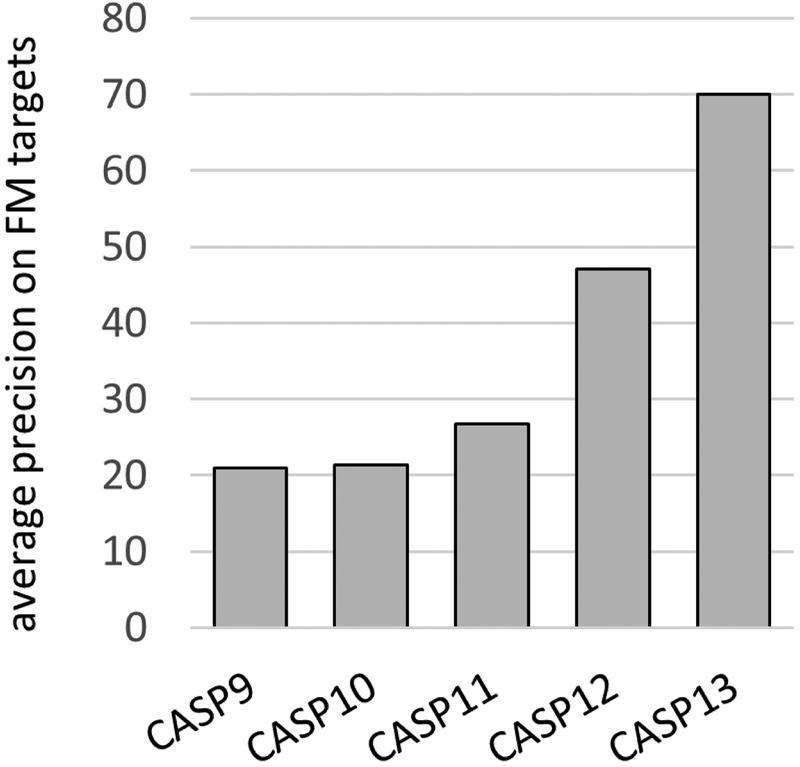
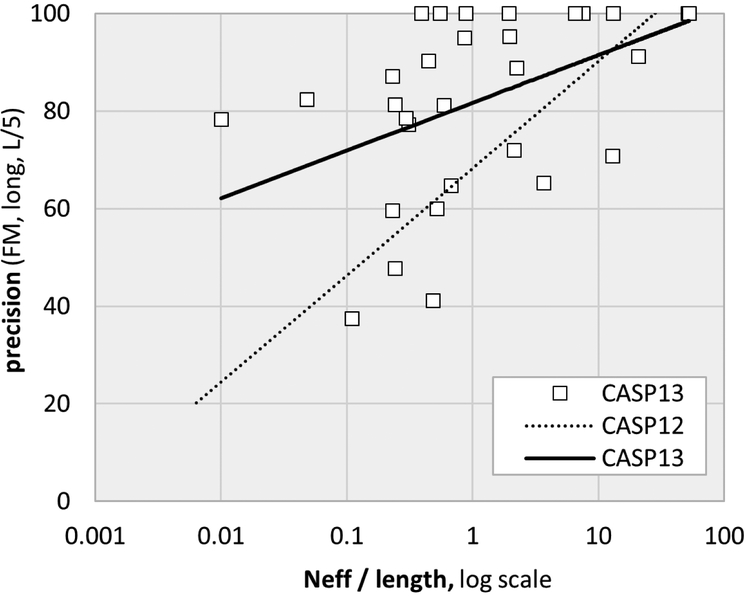
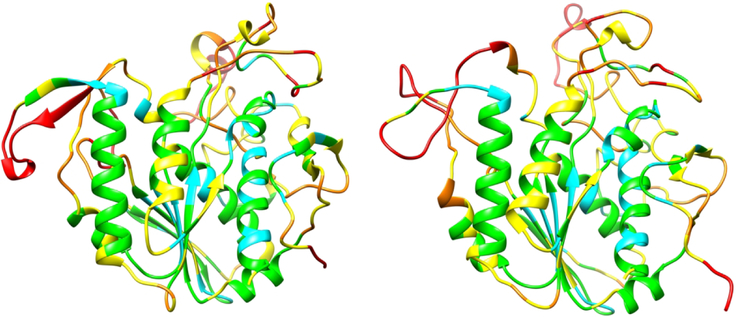
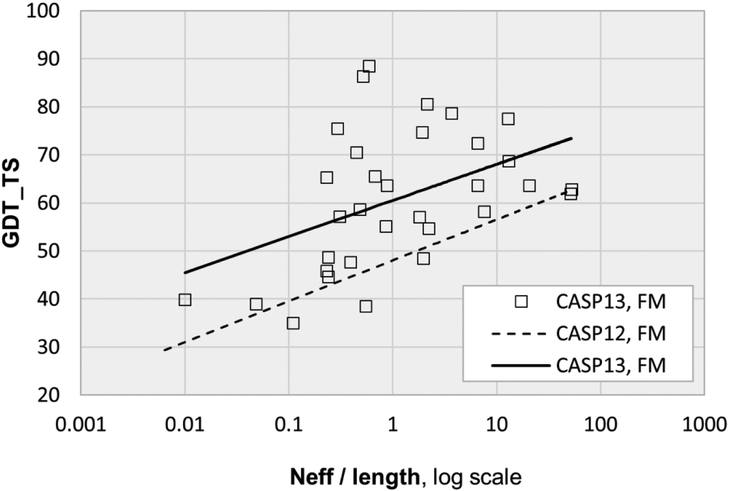
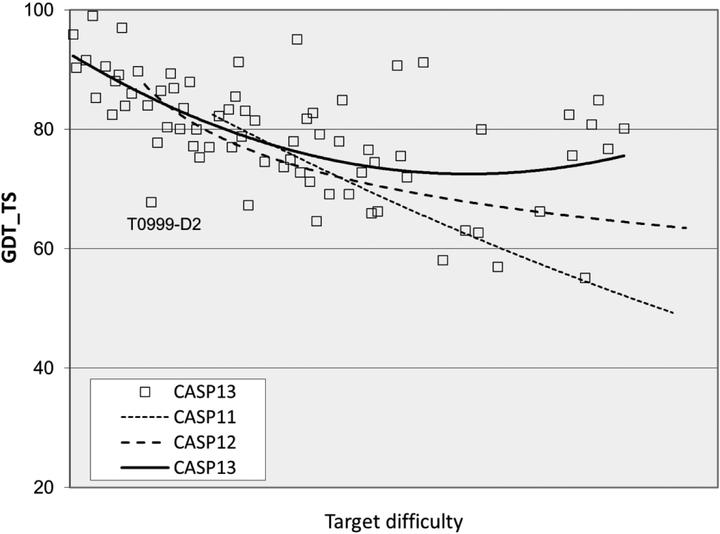
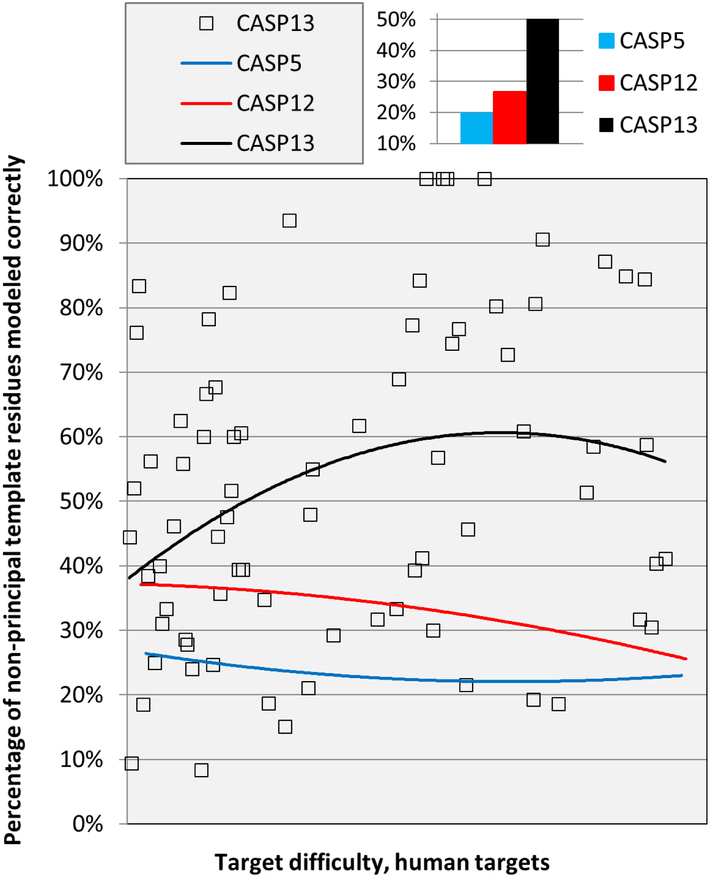
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
