

Exploring single-cell data with deep multitasking neural networks
- PMID: 31591579
- PMCID: PMC10164410
- DOI: 10.1038/s41592-019-0576-7
Exploring single-cell data with deep multitasking neural networks
Abstract
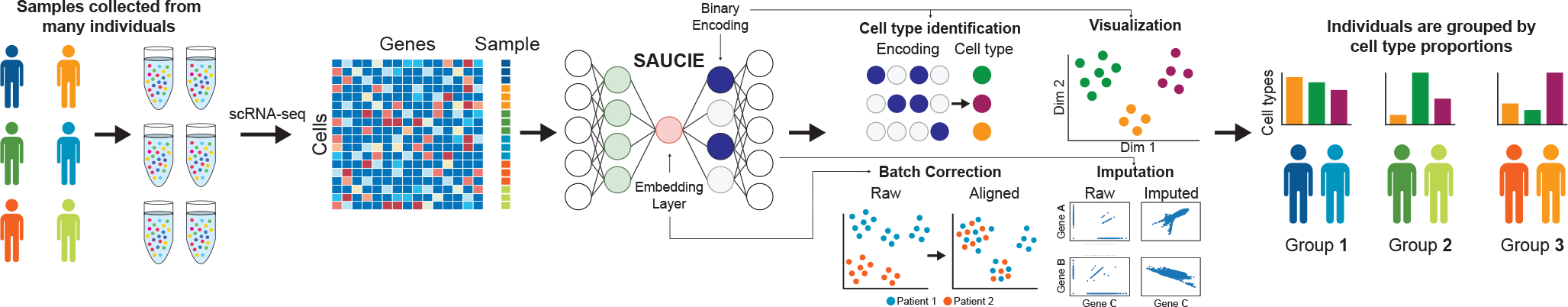
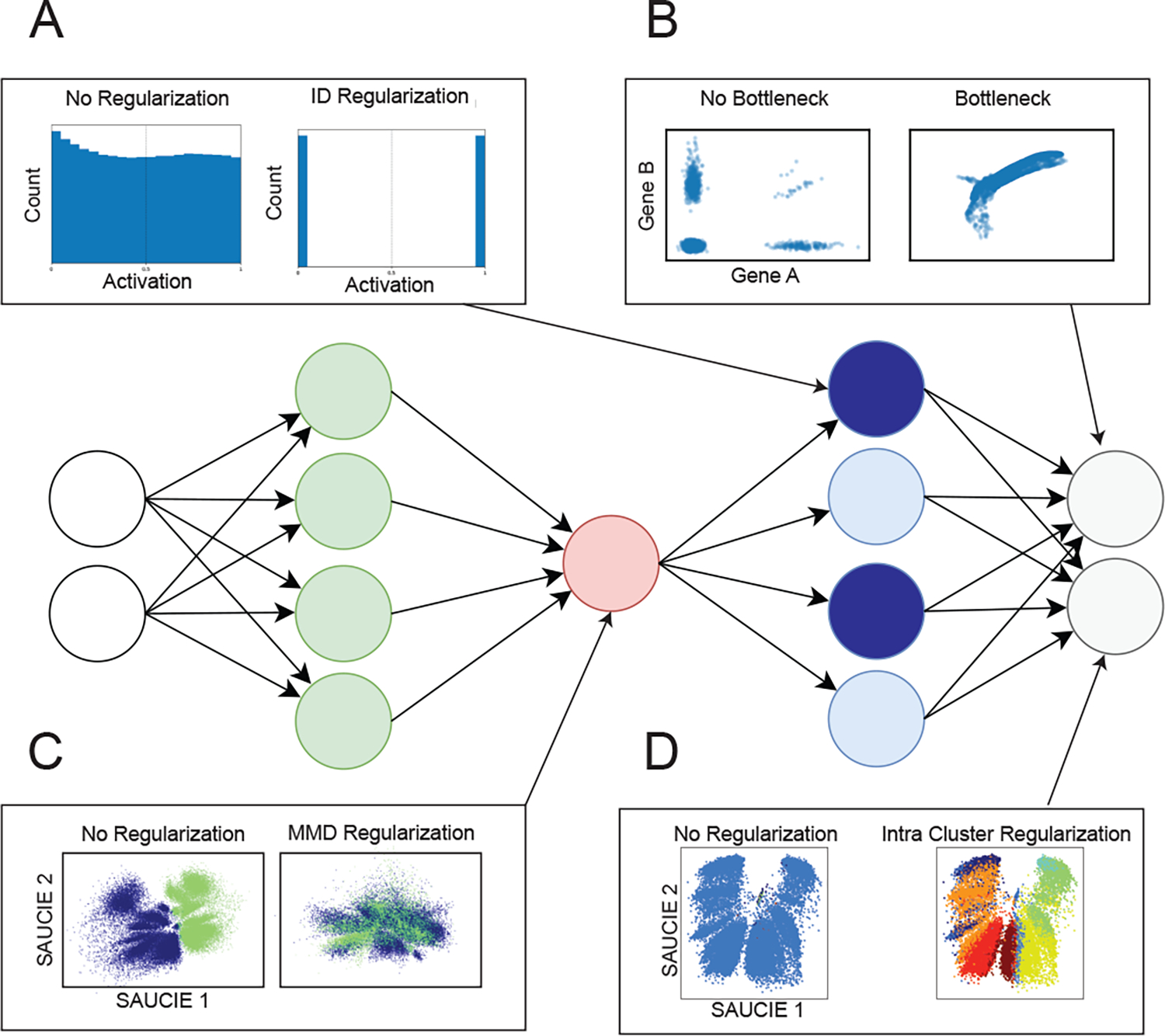
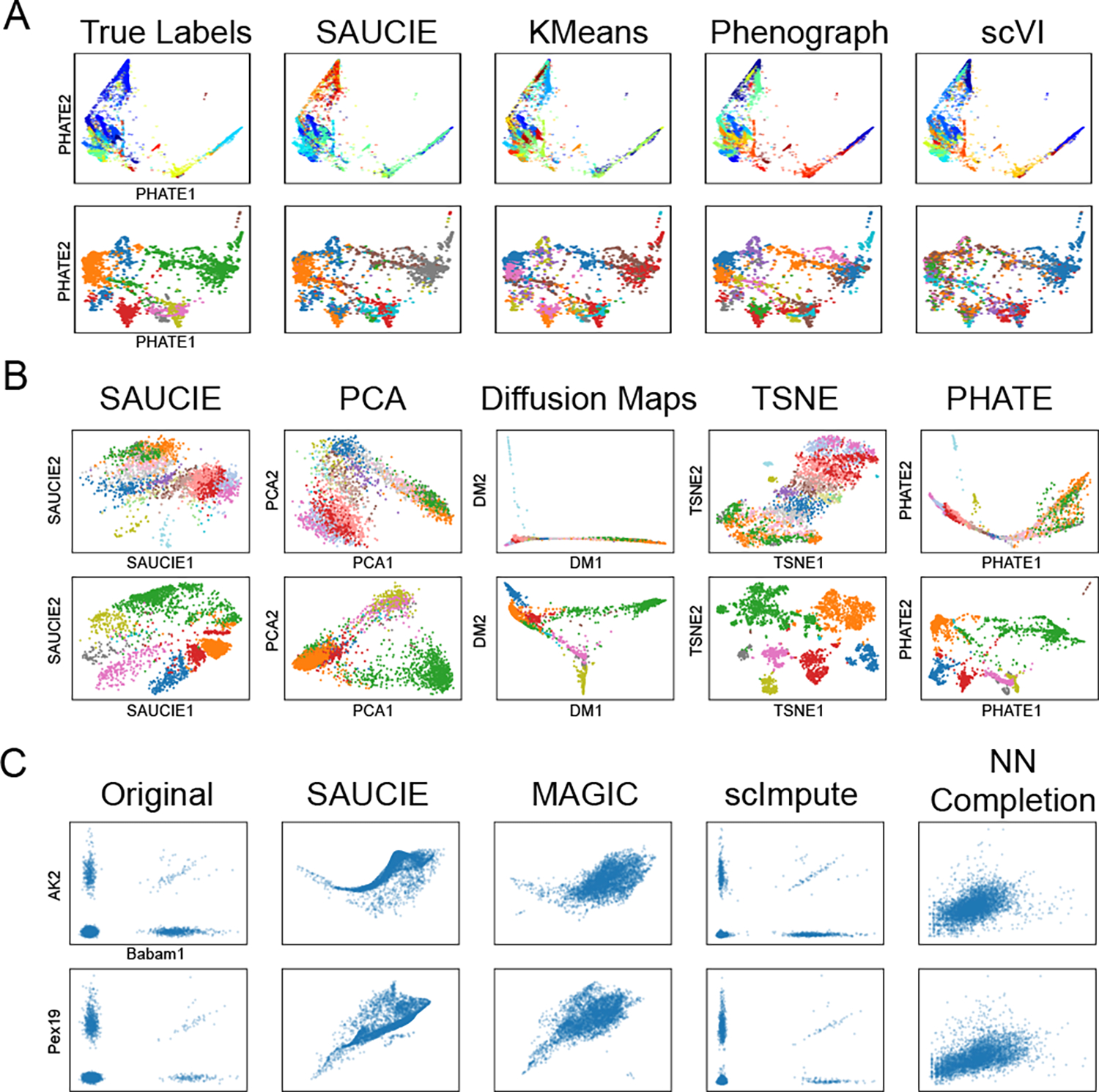
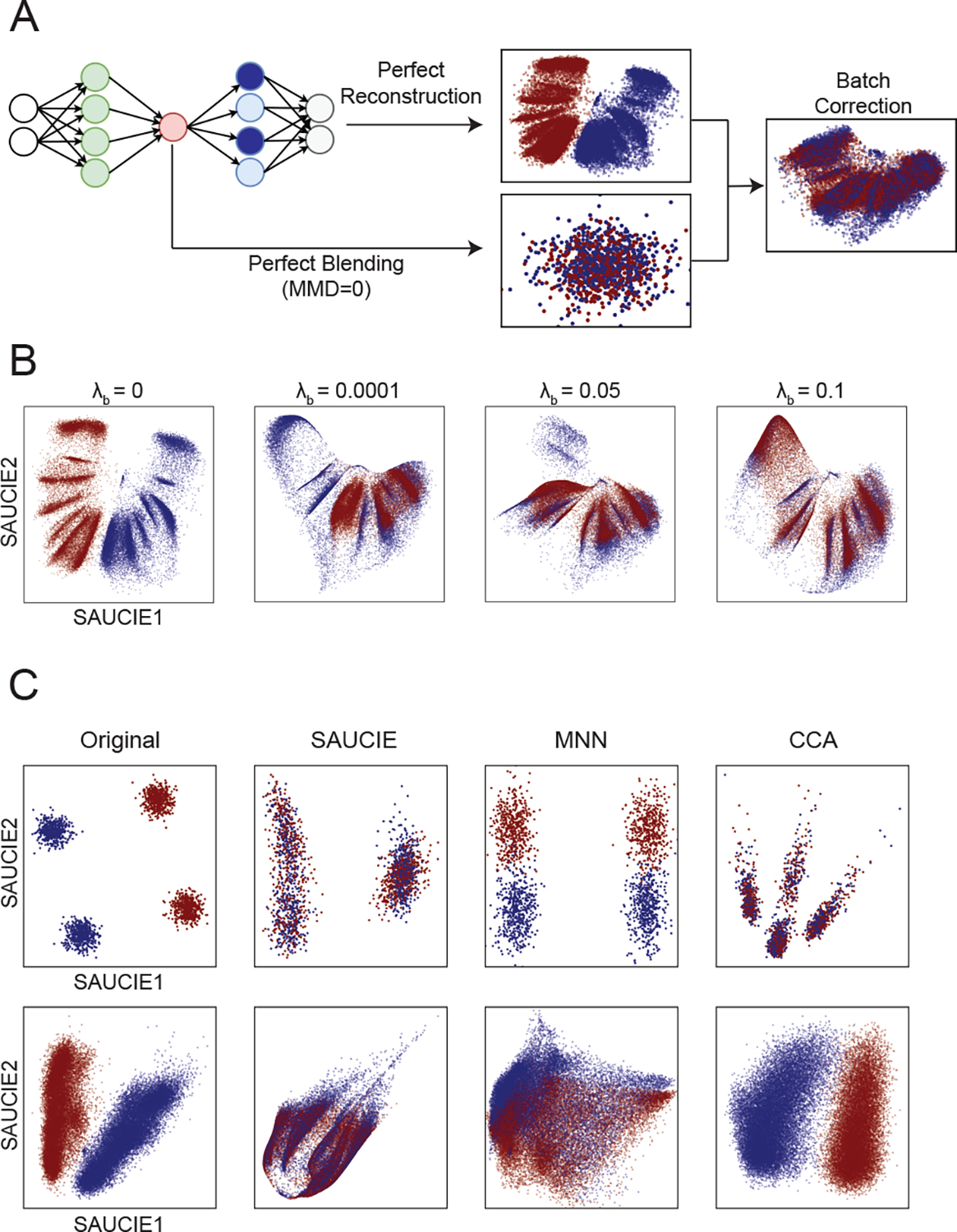
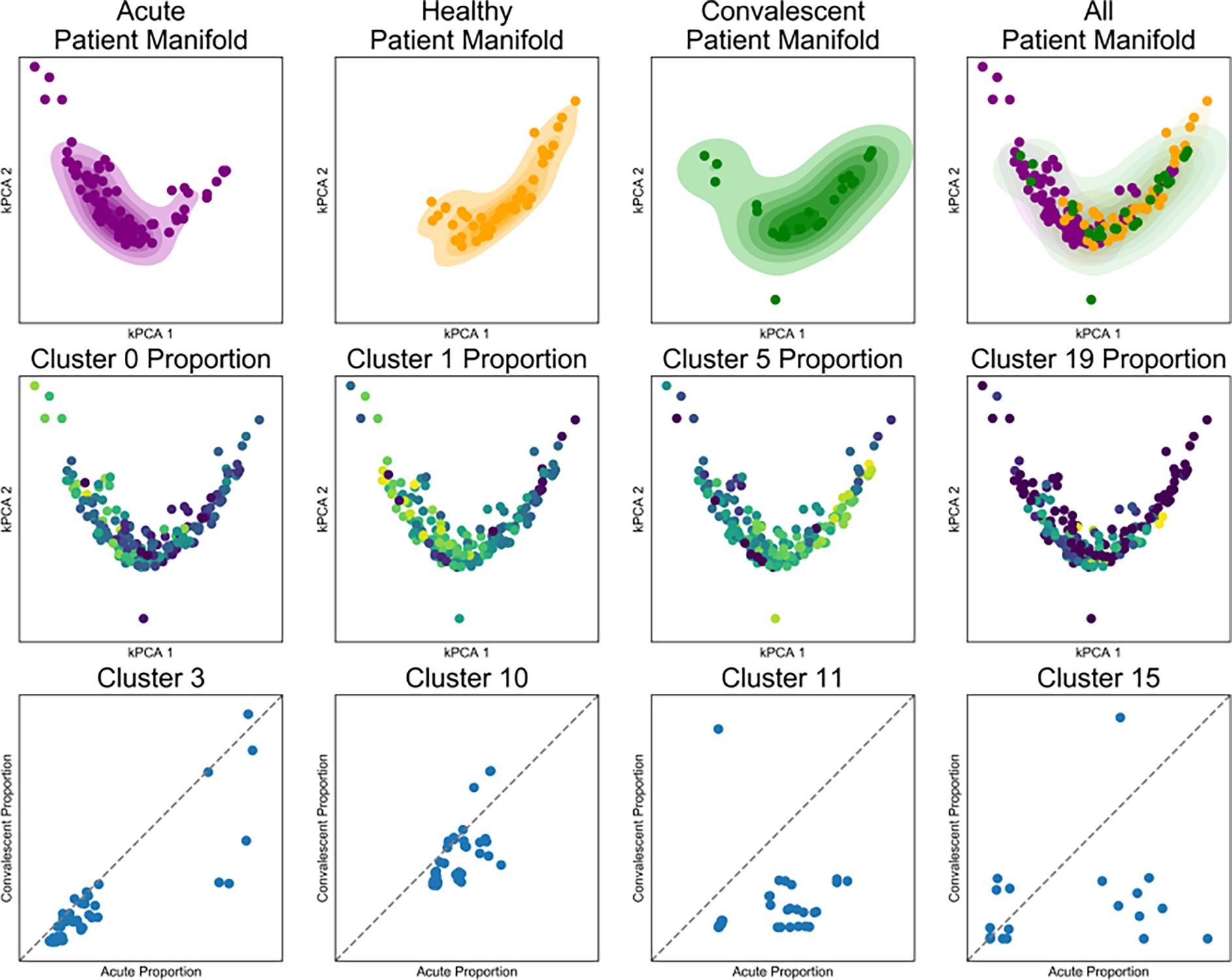
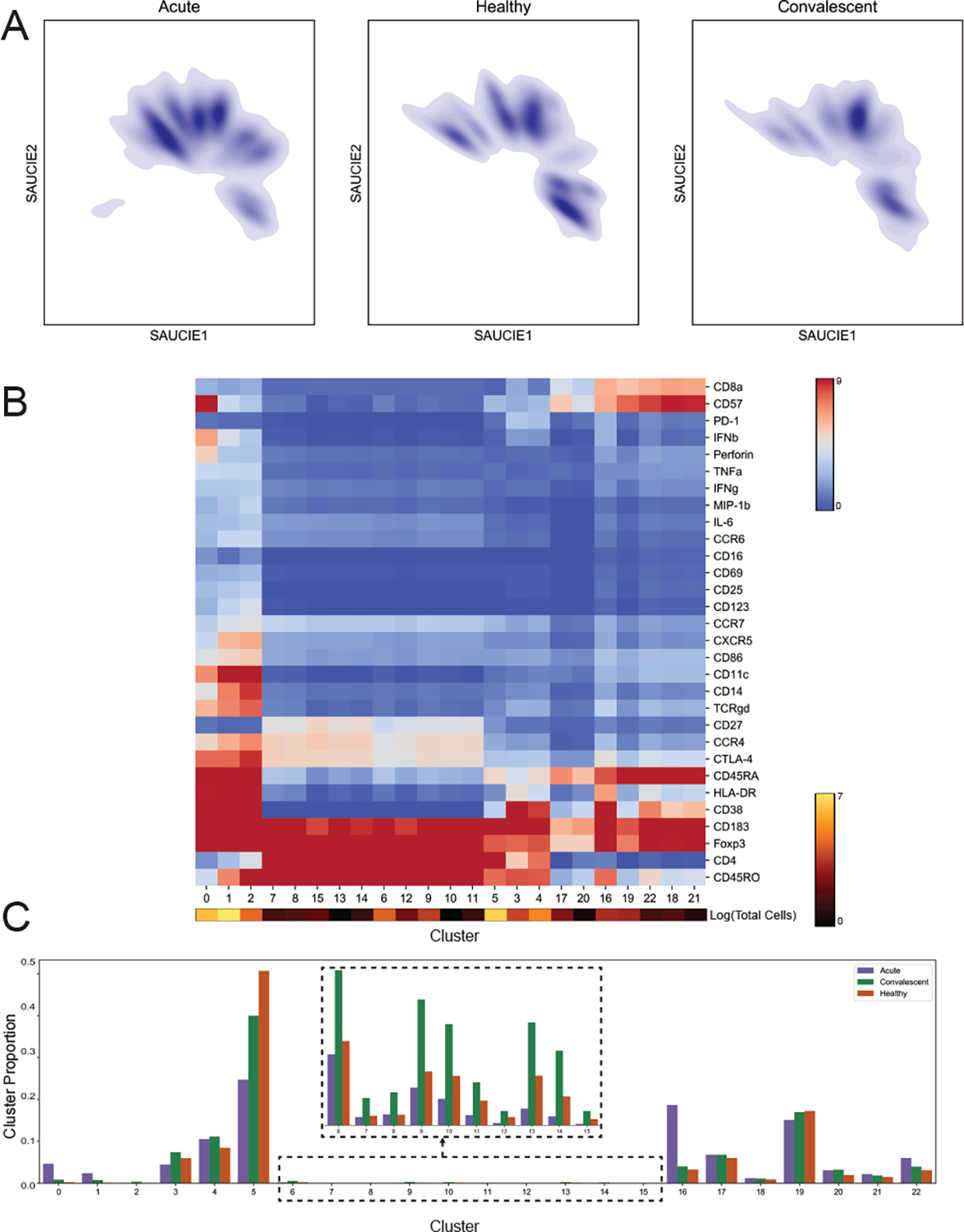
It is currently challenging to analyze single-cell data consisting of many cells and samples, and to address variations arising from batch effects and different sample preparations. For this purpose, we present SAUCIE, a deep neural network that combines parallelization and scalability offered by neural networks, with the deep representation of data that can be learned by them to perform many single-cell data analysis tasks. Our regularizations (penalties) render features learned in hidden layers of the neural network interpretable. On large, multi-patient datasets, SAUCIE's various hidden layers contain denoised and batch-corrected data, a low-dimensional visualization and unsupervised clustering, as well as other information that can be used to explore the data. We analyze a 180-sample dataset consisting of 11 million T cells from dengue patients in India, measured with mass cytometry. SAUCIE can batch correct and identify cluster-based signatures of acute dengue infection and create a patient manifold, stratifying immune response to dengue.
Conflict of interest statement
Competing Financial Interests Statement
There are no competing interests.
Figures
References
-
- Wang W, Huang Y, Wang Y, and Wang L, “Generalized autoencoder: A neural network framework for dimensionality reduction,” in CVPR Workshops, 2014.
Methods-only References
-
- Moon KR, Stanley J, Burkhardt D, van Dijk D, Wolf G, and Krishnaswamy S, “Manifold learning-based methods for analyzing single-cell RNA-sequencing data,” Current Opinion in Systems Biology, 2017.
-
- Moon KR, van Dijk D, Wang Z, Chen W, Hirn MJ, Coifman RR, Ivanova NB, Wolf G, and Krishnaswamy S, “PHATE: A dimensionality reduction method for visualizing trajectory structures in high-dimensional biological data,” bioRxiv, p. 120378, 2017.
-
- Montufar GF, Pascanu R, Cho K, and Bengio Y, “On the number of linear regions of deep neural networks,” in Advances in neural information processing systems, pp. 2924–2932, 2014.
-
- Anand K, Bianconi G, and Severini S, “Shannon and von Neumann entropy of random networks with heterogeneous expected degree,” Physical Review E, vol. 83, no. 3, p. 036109, 2011. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
