

Genome-wide germline correlates of the epigenetic landscape of prostate cancer
- PMID: 31591588
- PMCID: PMC7418214
- DOI: 10.1038/s41591-019-0579-z
Genome-wide germline correlates of the epigenetic landscape of prostate cancer
Abstract
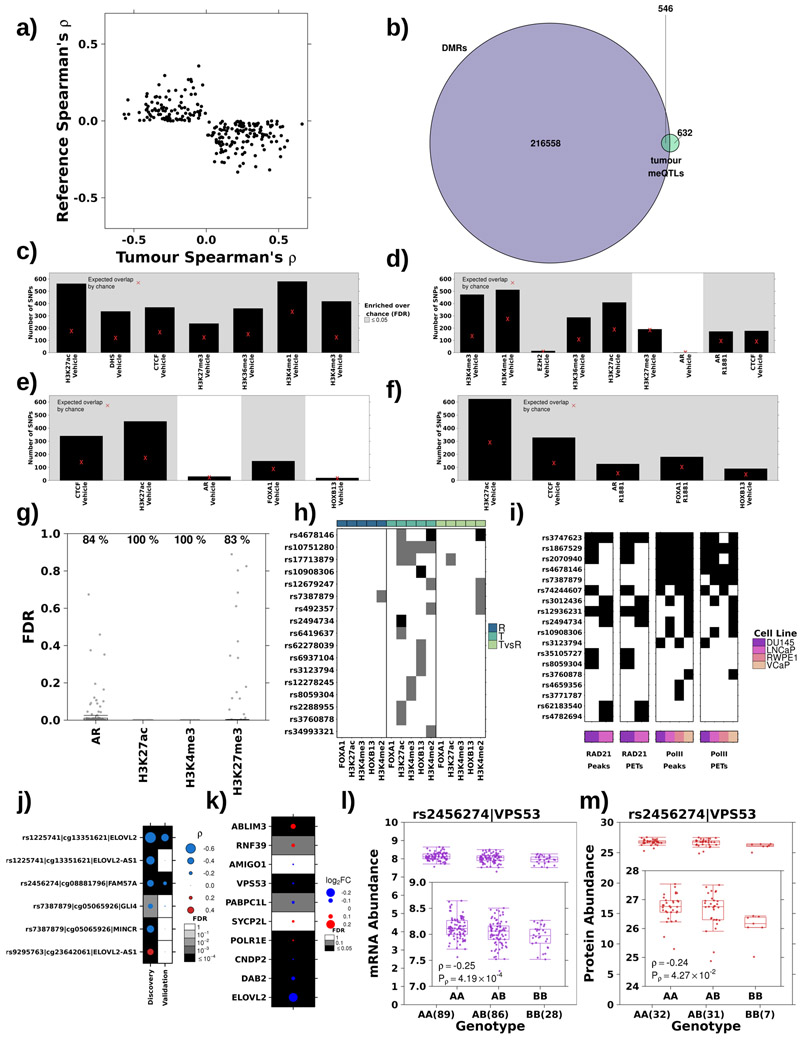
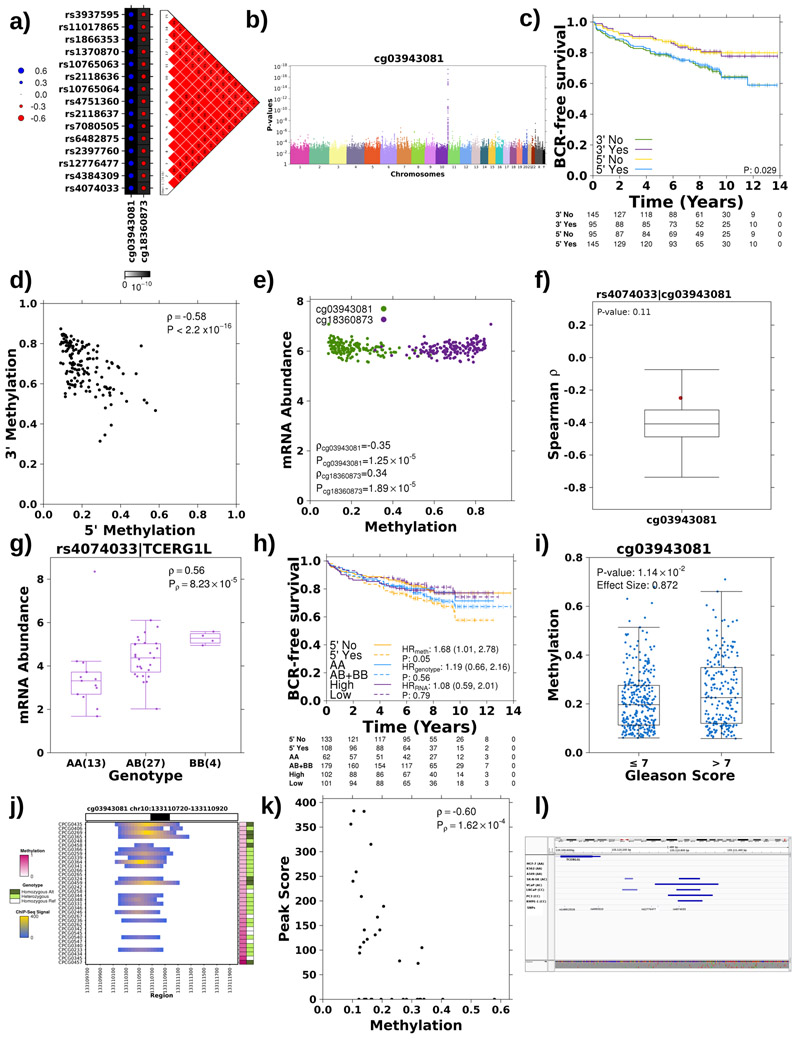
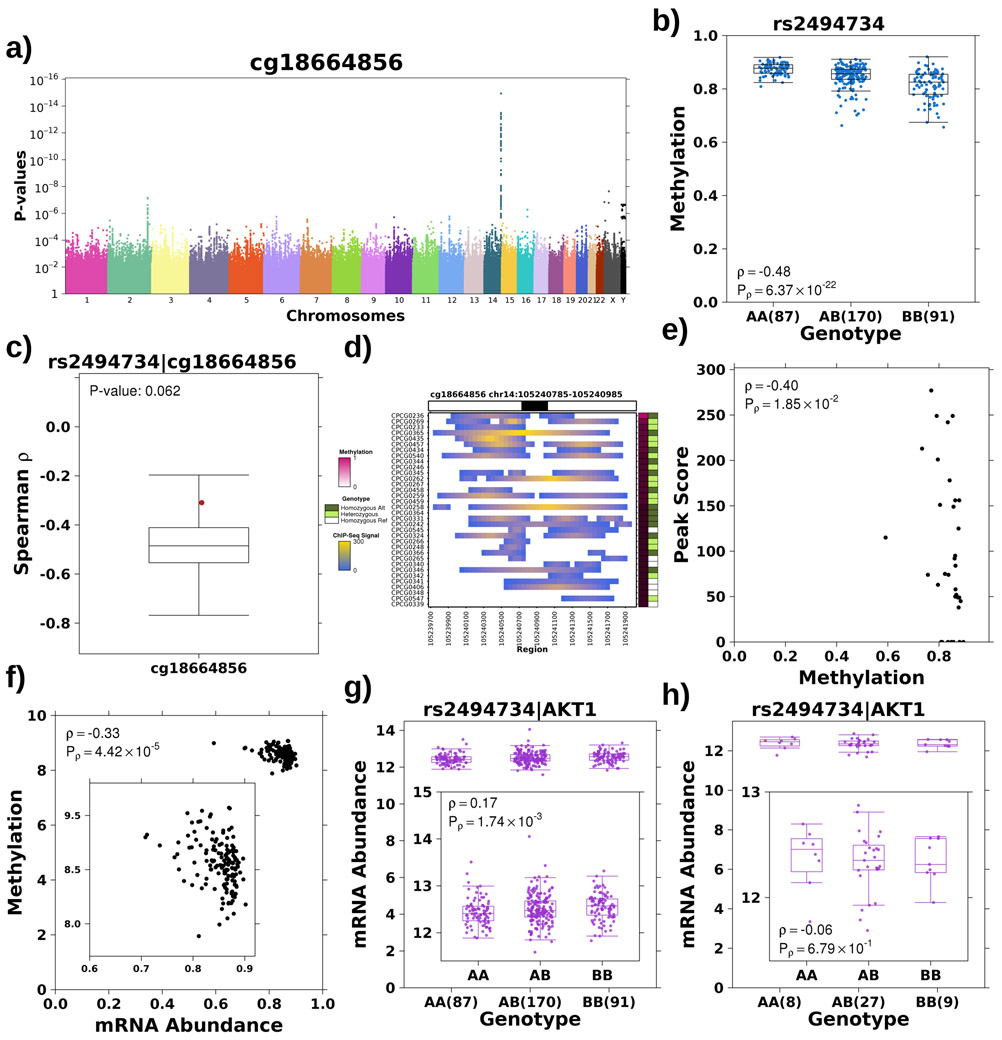
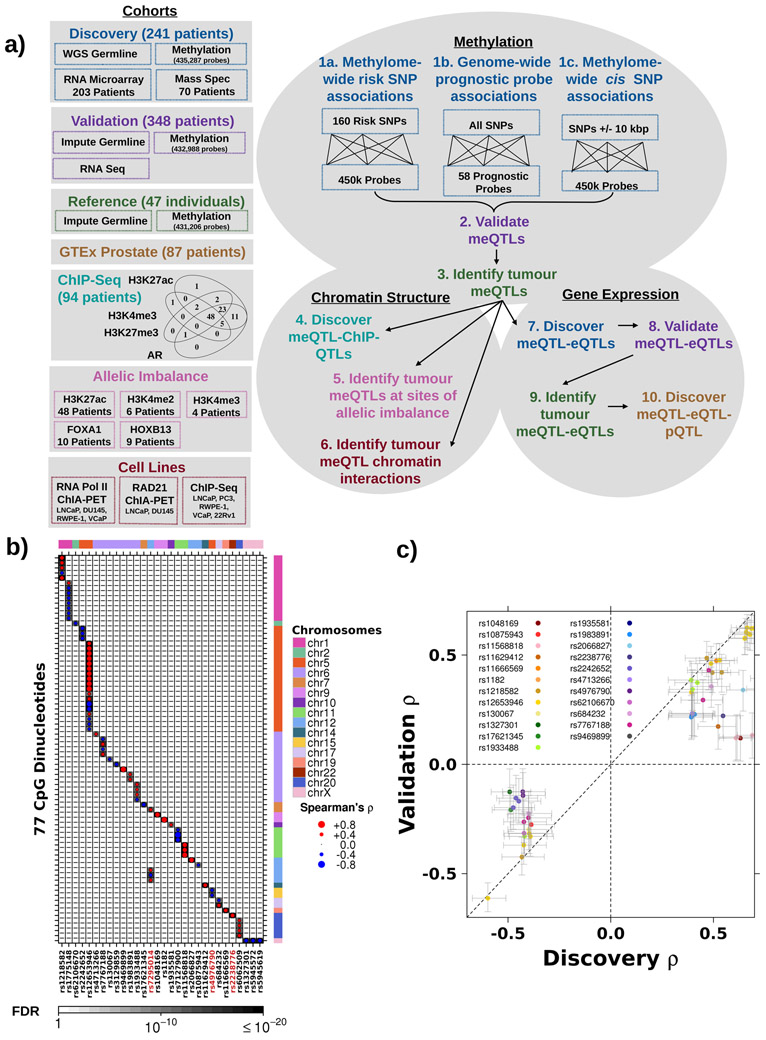
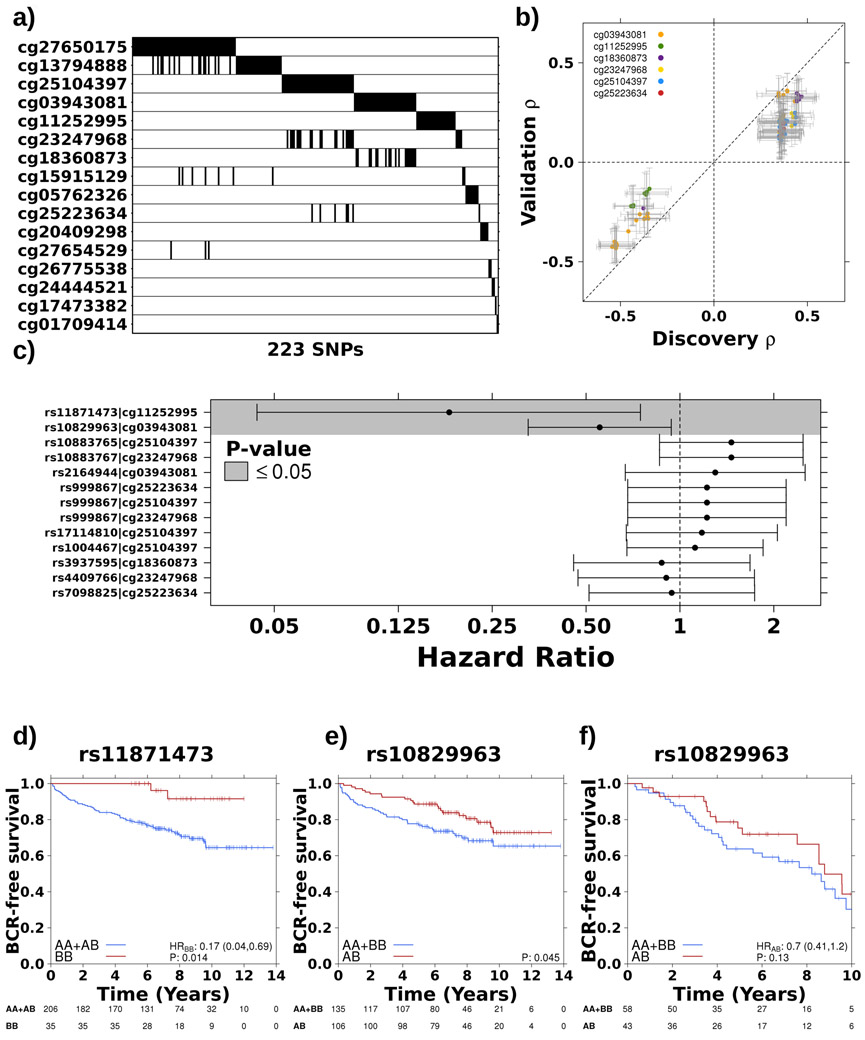
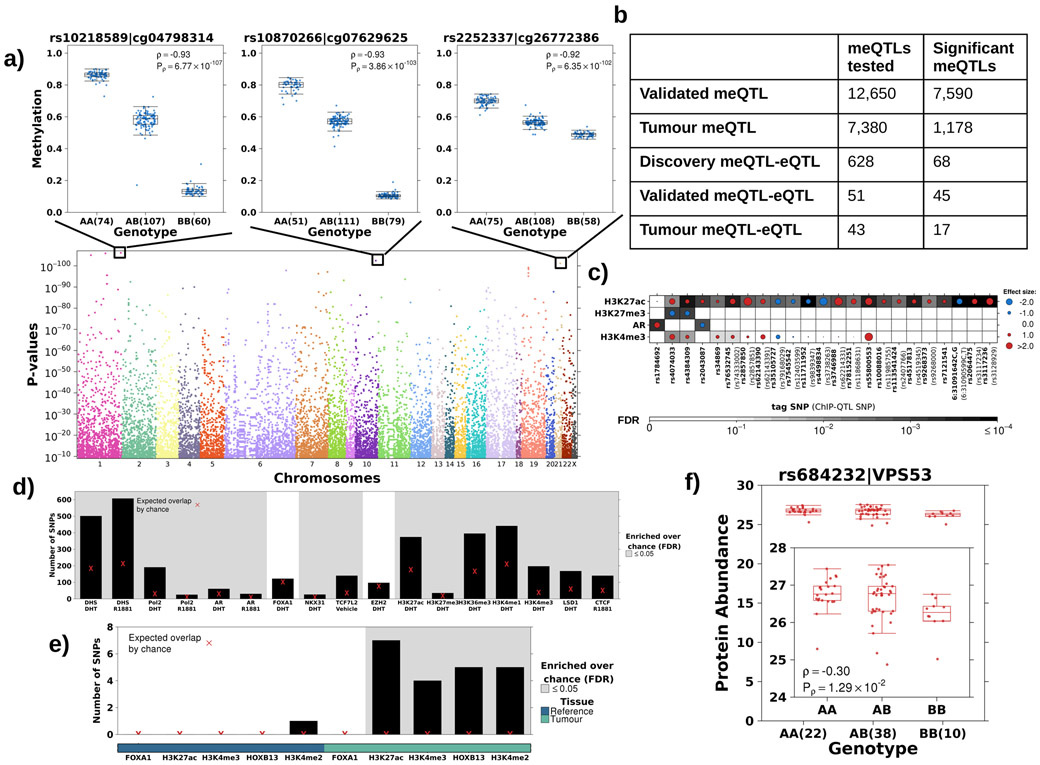
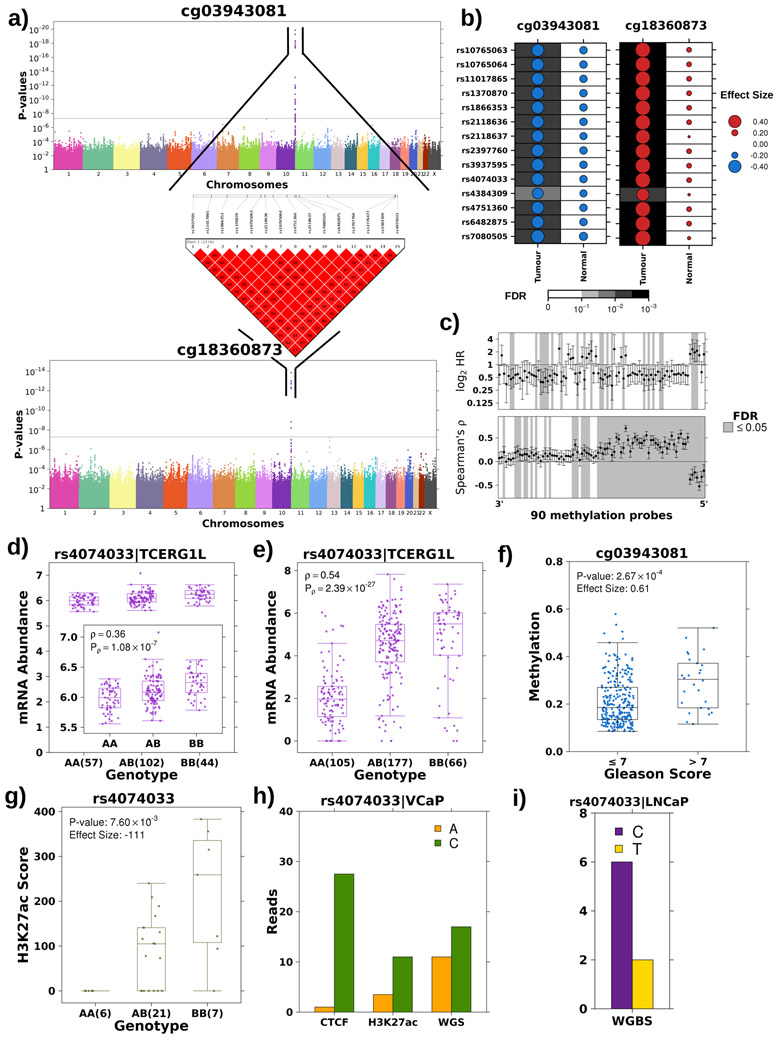
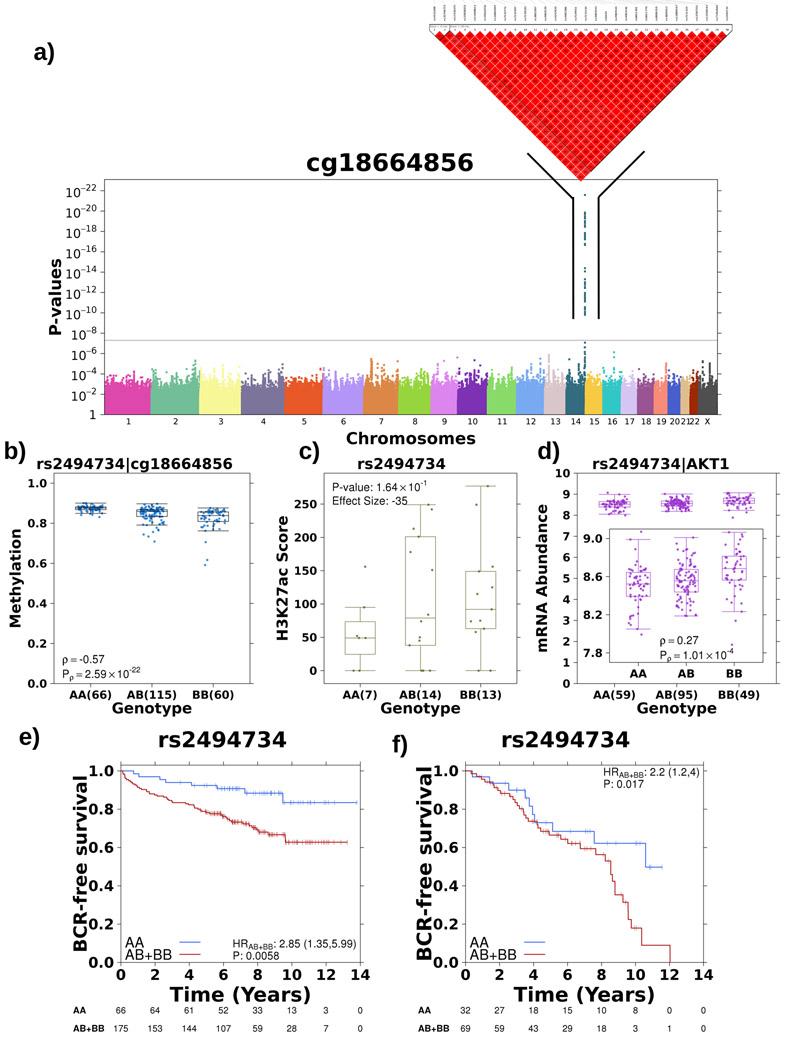
Oncogenesis is driven by germline, environmental and stochastic factors. It is unknown how these interact to produce the molecular phenotypes of tumors. We therefore quantified the influence of germline polymorphisms on the somatic epigenome of 589 localized prostate tumors. Predisposition risk loci influence a tumor's epigenome, uncovering a mechanism for cancer susceptibility. We identified and validated 1,178 loci associated with altered methylation in tumoral but not nonmalignant tissue. These tumor methylation quantitative trait loci influence chromatin structure, as well as RNA and protein abundance. One prominent tumor methylation quantitative trait locus is associated with AKT1 expression and is predictive of relapse after definitive local therapy in both discovery and validation cohorts. These data reveal intricate crosstalk between the germ line and the epigenome of primary tumors, which may help identify germline biomarkers of aggressive disease to aid patient triage and optimize the use of more invasive or expensive diagnostic assays.
Conflict of interest statement
Conflict of Interest Statement
All authors declare that they have no conflicts of interest.
Figures
Comment in
-
Germline-epigenome crosstalk promotes aggressive subsets of prostate tumours.Nat Rev Urol. 2019 Dec;16(12):693. doi: 10.1038/s41585-019-0254-7. Nat Rev Urol. 2019. PMID: 31659333 No abstract available.
References
-
- Hanahan D & Weinberg RA Hallmarks of cancer: The next generation. Cell 144, 646–674 (2011). - PubMed
-
- Garraway LA & Lander ES Lessons from the cancer genome. Cell 153, 17–37 (2013). - PubMed
-
- Tomlinson IP et al. A genome-wide association study identifies colorectal cancer susceptibility loci on chromosomes 10p14 and 8q23.3. Nat. Genet 40, 623–630 (2008). - PubMed
Methods References
-
- Shiah Y-J, Fraser M, Bristow RG & Boutros PC Comparison of Pre-processing Methods for Infinium HumanMethylation450 BeadChip Array. Bioinformatics 33, 3151–3157 (2017). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
