

A clonal expression biomarker associates with lung cancer mortality
- PMID: 31591602
- PMCID: PMC6984959
- DOI: 10.1038/s41591-019-0595-z
A clonal expression biomarker associates with lung cancer mortality
Erratum in
-
Publisher Correction: A clonal expression biomarker associates with lung cancer mortality.Nat Med. 2020 Jul;26(7):1148. doi: 10.1038/s41591-020-0899-z. Nat Med. 2020. PMID: 32494065
Abstract
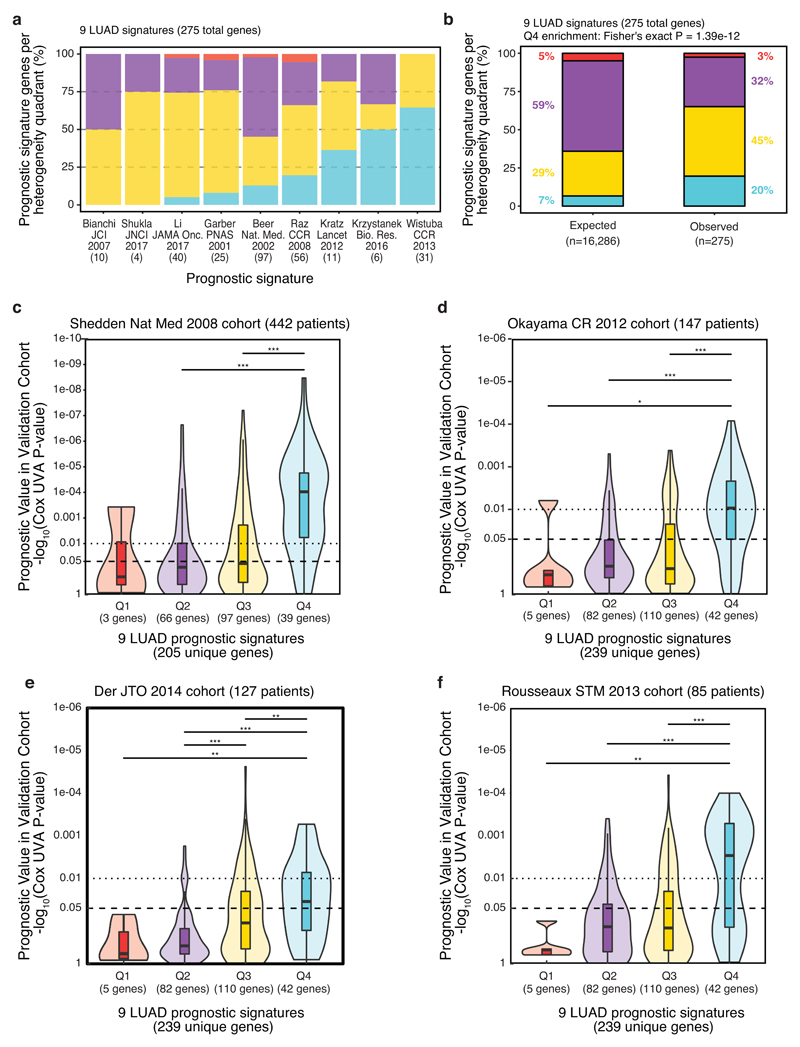
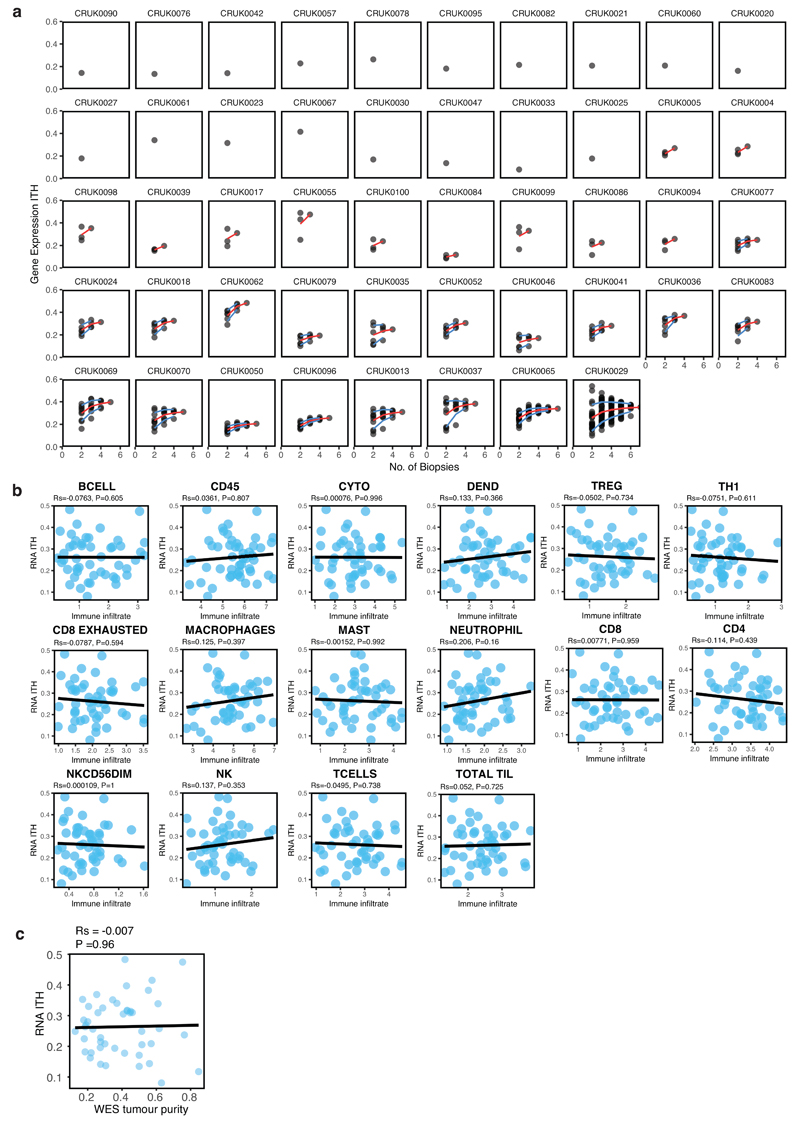
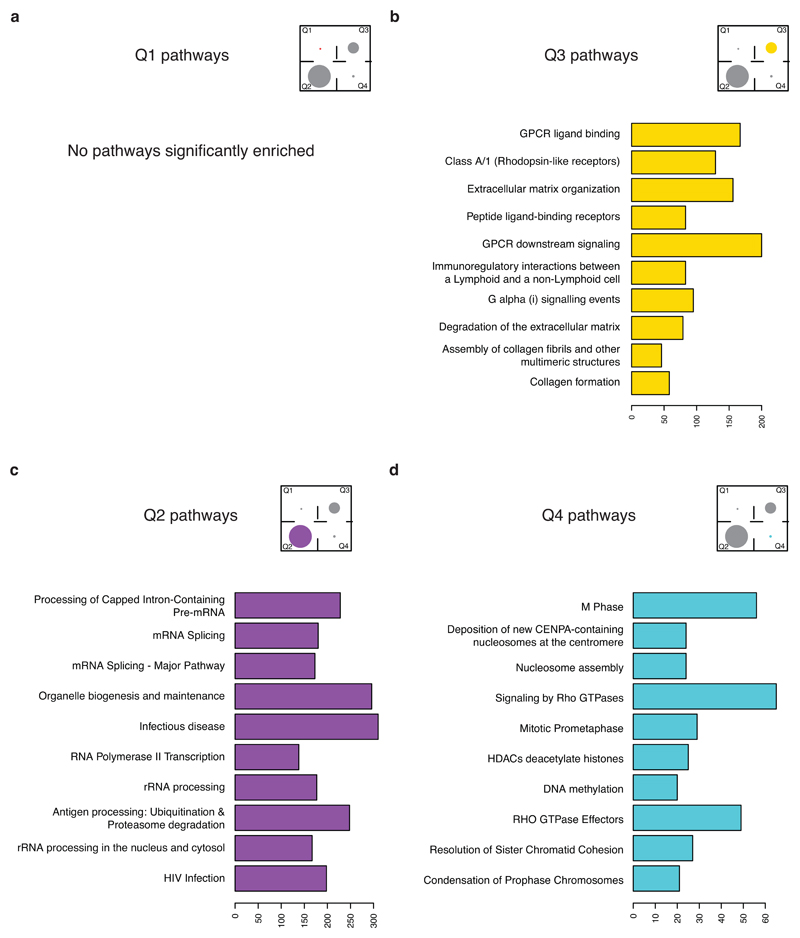
An aim of molecular biomarkers is to stratify patients with cancer into disease subtypes predictive of outcome, improving diagnostic precision beyond clinical descriptors such as tumor stage1. Transcriptomic intratumor heterogeneity (RNA-ITH) has been shown to confound existing expression-based biomarkers across multiple cancer types2-6. Here, we analyze multi-region whole-exome and RNA sequencing data for 156 tumor regions from 48 patients enrolled in the TRACERx study to explore and control for RNA-ITH in non-small cell lung cancer. We find that chromosomal instability is a major driver of RNA-ITH, and existing prognostic gene expression signatures are vulnerable to tumor sampling bias. To address this, we identify genes expressed homogeneously within individual tumors that encode expression modules of cancer cell proliferation and are often driven by DNA copy-number gains selected early in tumor evolution. Clonal transcriptomic biomarkers overcome tumor sampling bias, associate with survival independent of clinicopathological risk factors, and may provide a general strategy to refine biomarker design across cancer types.
Conflict of interest statement
C.S. receives grant support from Pfizer, AstraZeneca, BMS, Roche and Ventana. C.S. has consulted for Pfizer, Novartis, GlaxoSmithKline, MSD, BMS, Celgene, AstraZeneca, Illumina, Genentech, Roche-Ventana, GRAIL, Medicxi, the Sarah Cannon Research Institute and is an Advisor for Dynamo Therapeutics. C.S. is a shareholder of Apogen Biotechnologies, Epic Bioscience, GRAIL, and has stock options in and is co-founder of Achilles Therapeutics. R.R. has stock options in and has consulted for Achilles Therapeutics. C.A. has received speaking honorarium or expenses from Novartis, Roche, AstraZeneca and BMS. M.A.B. has consulted for Achilles Therapeutics. G.A.W. is a shareholder of Achilles Therapeutics. M.J.-H. has consulted for and is an advisor for Achilles Therapeutics. D.B., N.J.B., N.M., and C.S. are co-inventors on a UK patent application (1901439.8) filed by Cancer Research Technology relating to methods of predicting survival rates for cancer patients.
Figures











Comment in
-
Tracing evolution reveals new biomarkers.Nat Rev Clin Oncol. 2020 Jan;17(1):5. doi: 10.1038/s41571-019-0295-0. Nat Rev Clin Oncol. 2020. PMID: 31645685 No abstract available.
-
Re: A Clonal Expression Biomarker Associates with Lung Cancer Mortality.Eur Urol. 2020 Dec;78(6):925-926. doi: 10.1016/j.eururo.2020.06.027. Epub 2020 Jul 4. Eur Urol. 2020. PMID: 32636096 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
- FC001169/MRC_/Medical Research Council/United Kingdom
- 20265/CRUK_/Cancer Research UK/United Kingdom
- FC001169/CRUK_/Cancer Research UK/United Kingdom
- 19310/CRUK_/Cancer Research UK/United Kingdom
- FC001202/ARC_/Arthritis Research UK/United Kingdom
- 28990/CRUK_/Cancer Research UK/United Kingdom
- 211179/WT_/Wellcome Trust/United Kingdom
- FC001169/ARC_/Arthritis Research UK/United Kingdom
- 24956/CRUK_/Cancer Research UK/United Kingdom
- 17786/CRUK_/Cancer Research UK/United Kingdom
- 30025/CRUK_/Cancer Research UK/United Kingdom
- MC_UP_1203/1/MRC_/Medical Research Council/United Kingdom
- 21999/CRUK_/Cancer Research UK/United Kingdom
- 22246/CRUK_/Cancer Research UK/United Kingdom
- MR/P014712/1/MRC_/Medical Research Council/United Kingdom
- MR/M009033/1/MRC_/Medical Research Council/United Kingdom
- 25253/CRUK_/Cancer Research UK/United Kingdom
- 20466/CRUK_/Cancer Research UK/United Kingdom
- FC001169/WT_/Wellcome Trust/United Kingdom
- WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
