

Spatial heterogeneity of the T cell receptor repertoire reflects the mutational landscape in lung cancer
- PMID: 31591606
- PMCID: PMC6890490
- DOI: 10.1038/s41591-019-0592-2
Spatial heterogeneity of the T cell receptor repertoire reflects the mutational landscape in lung cancer
Erratum in
-
Publisher Correction: Spatial heterogeneity of the T cell receptor repertoire reflects the mutational landscape in lung cancer.Nat Med. 2020 Jul;26(7):1148. doi: 10.1038/s41591-020-0866-8. Nat Med. 2020. PMID: 32494063
Abstract
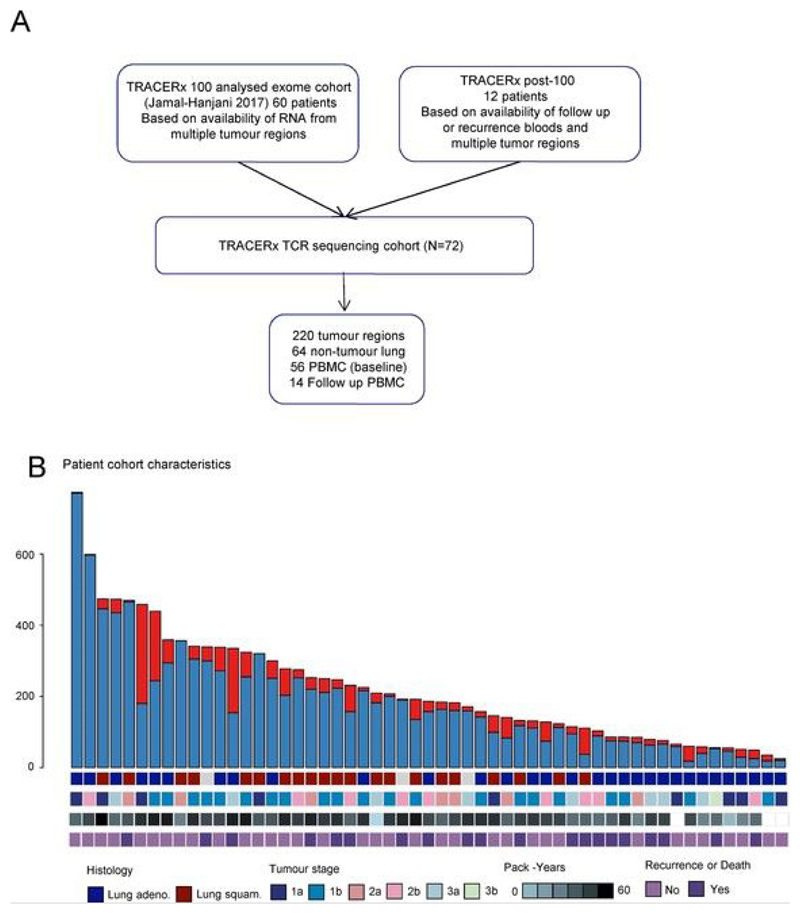
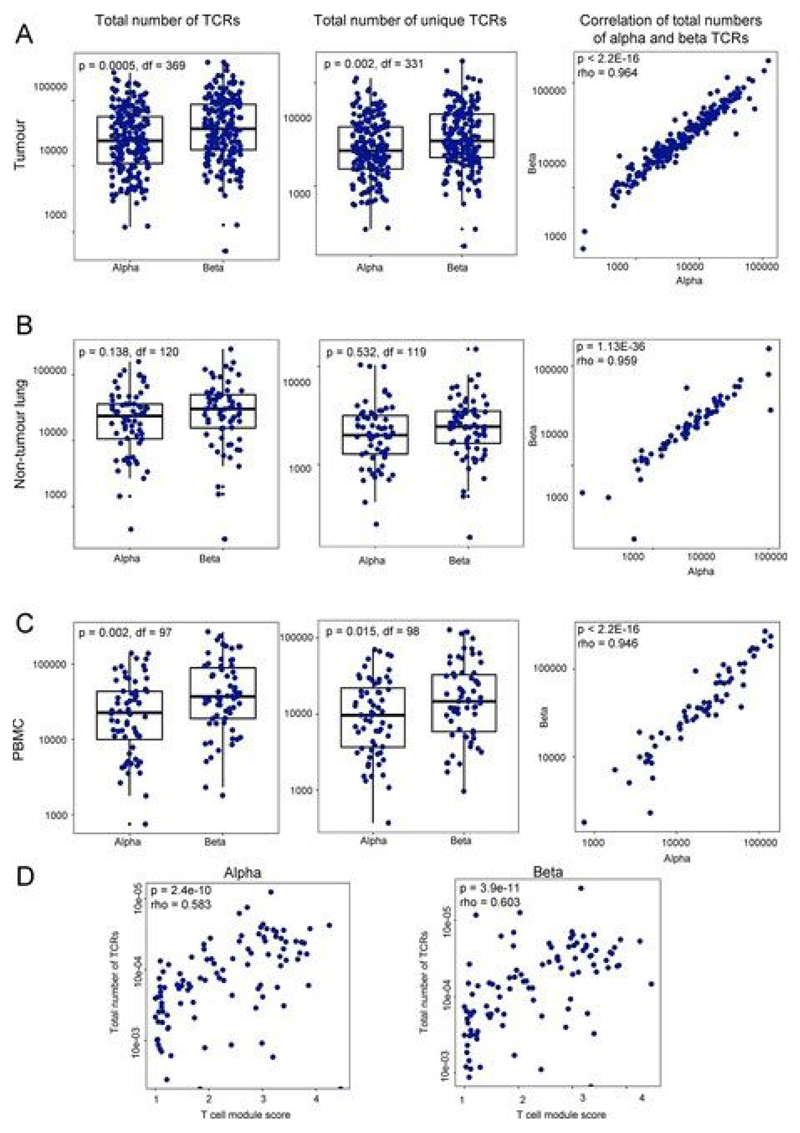
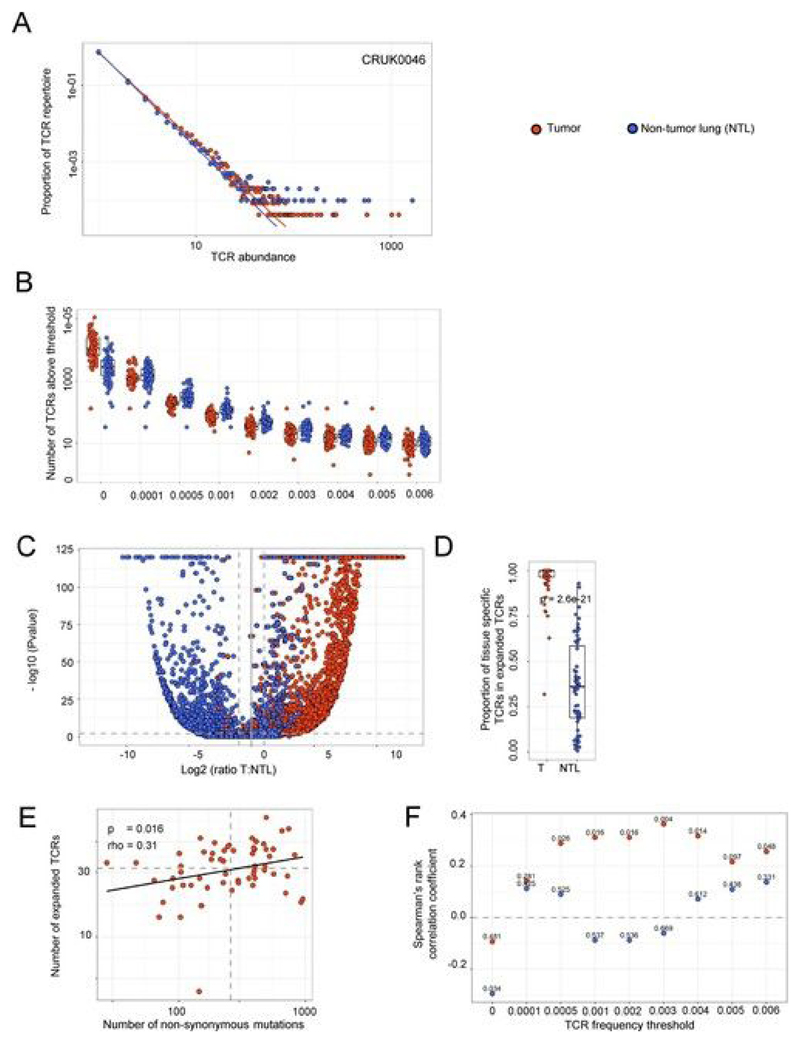
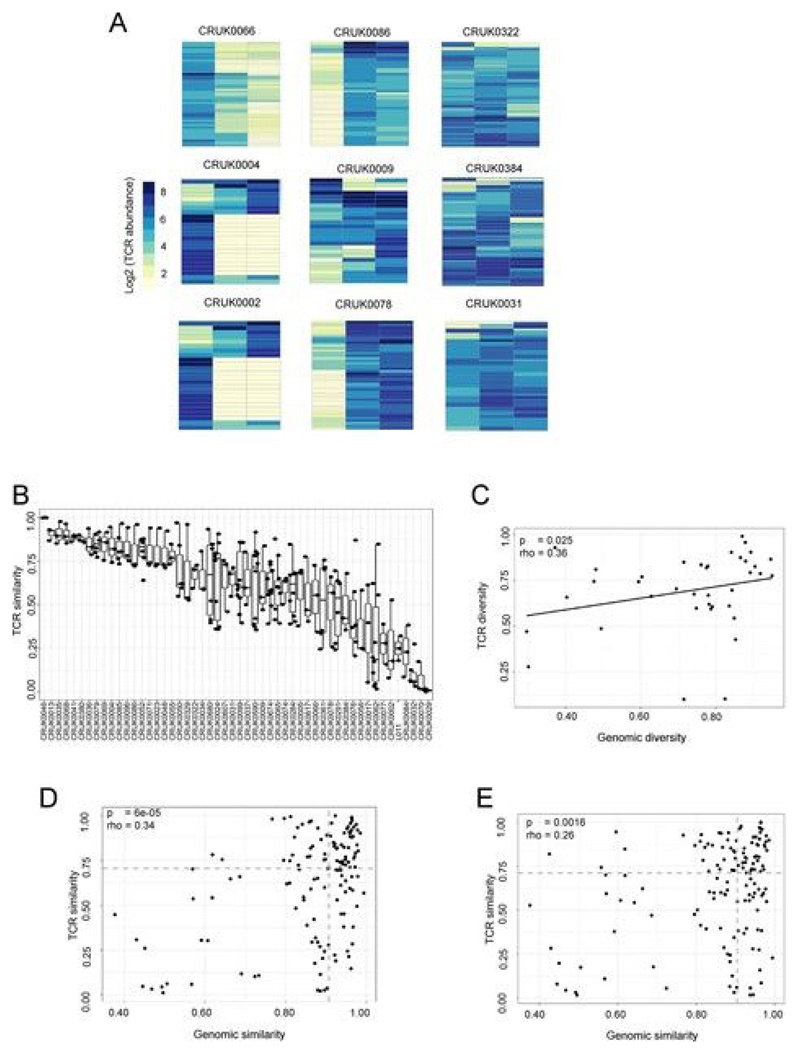
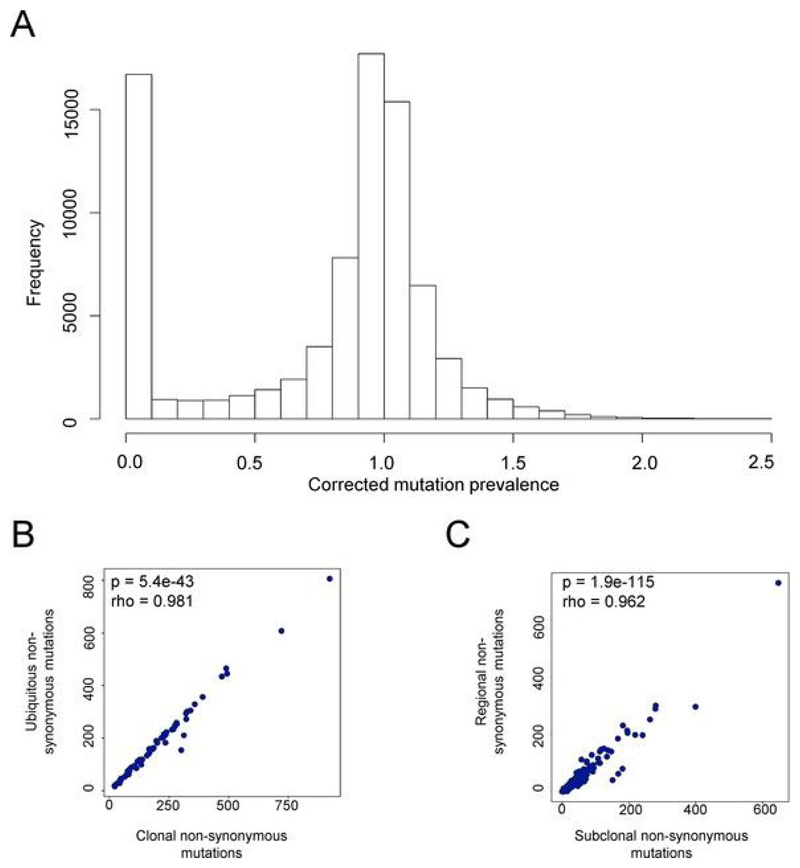
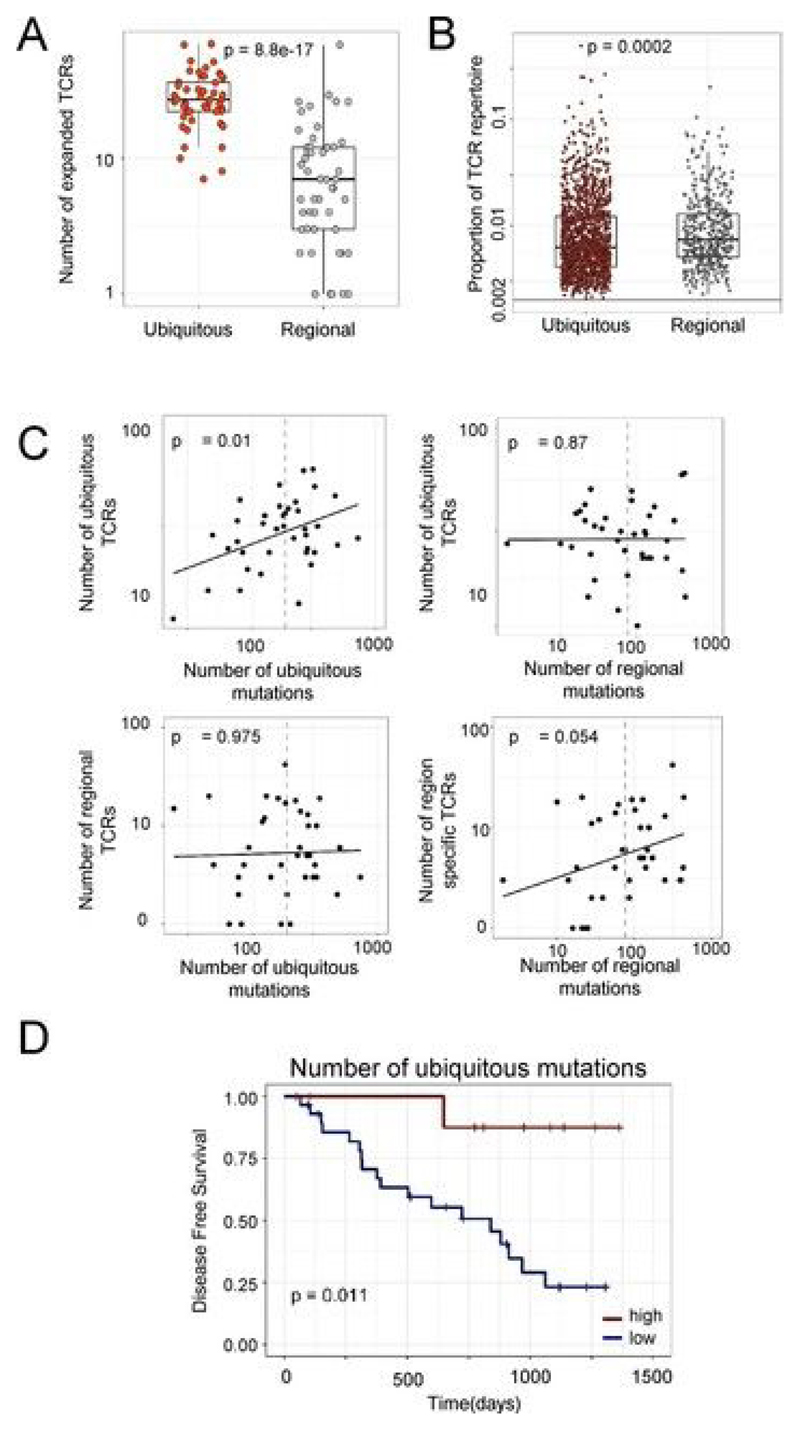
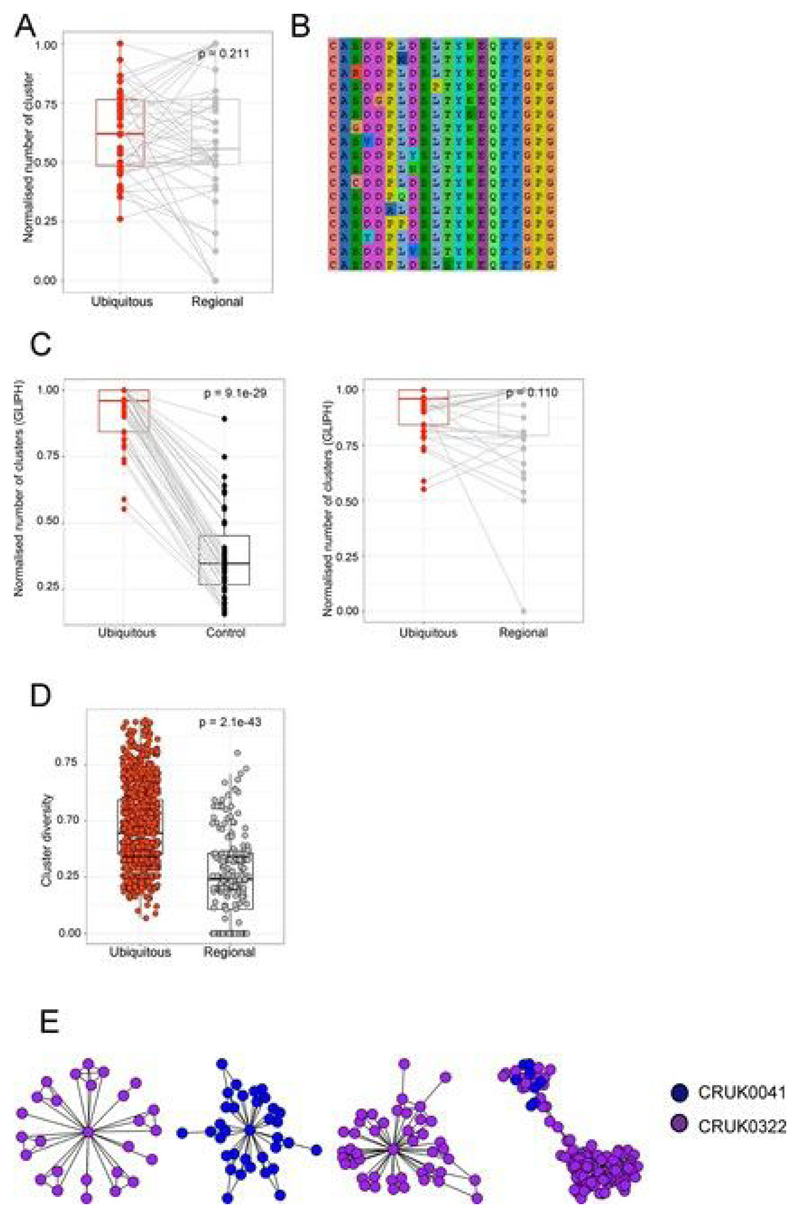
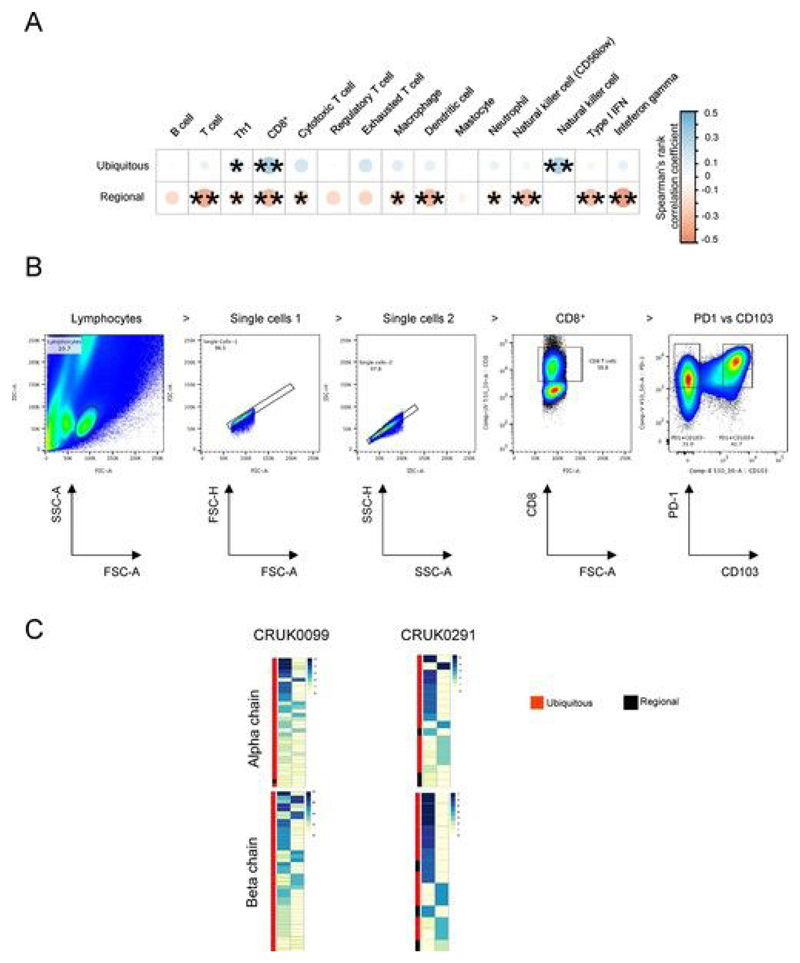
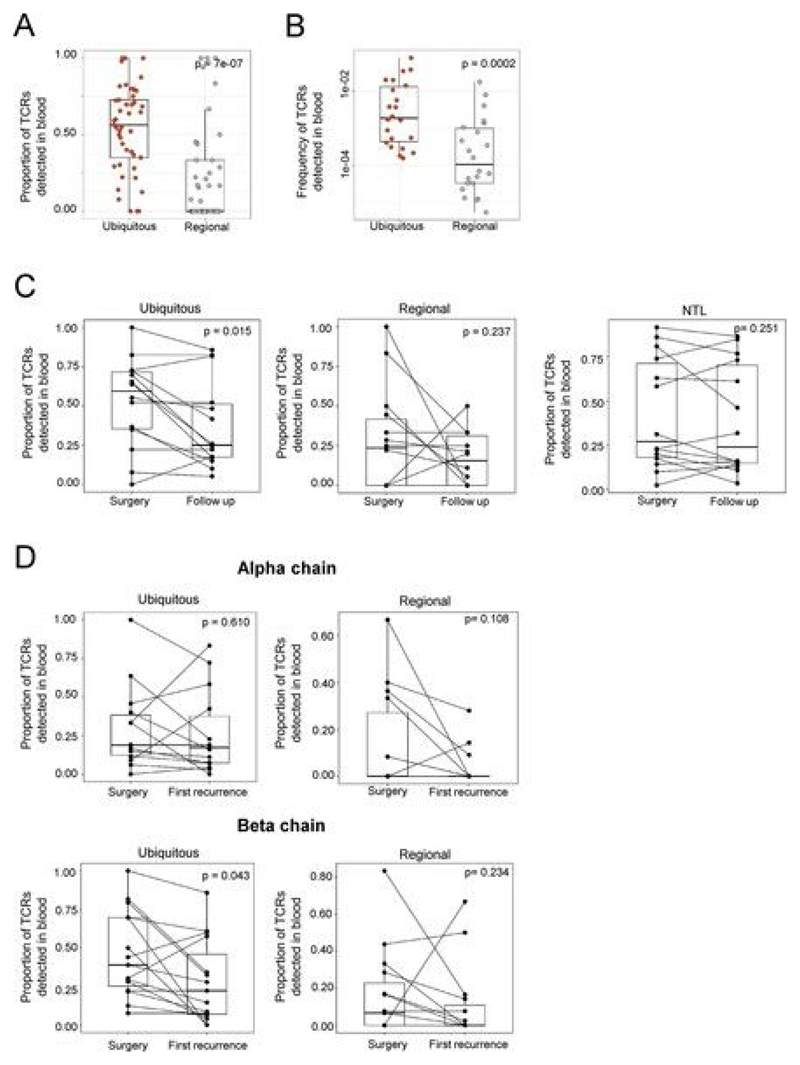
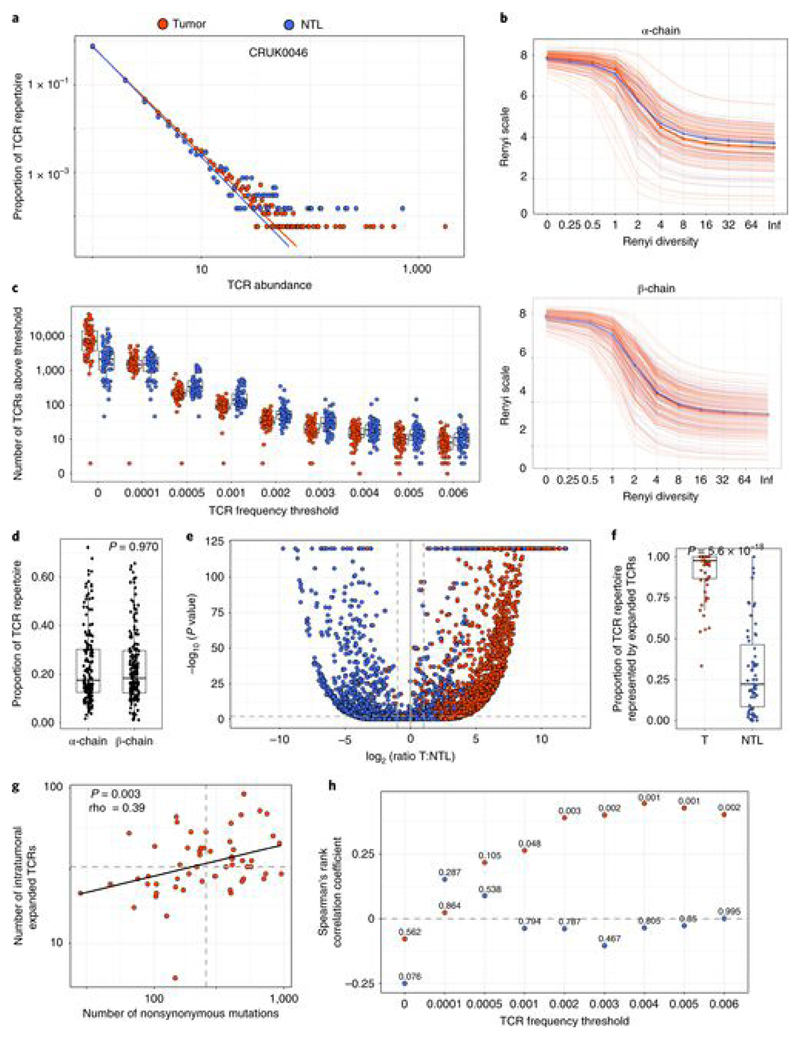
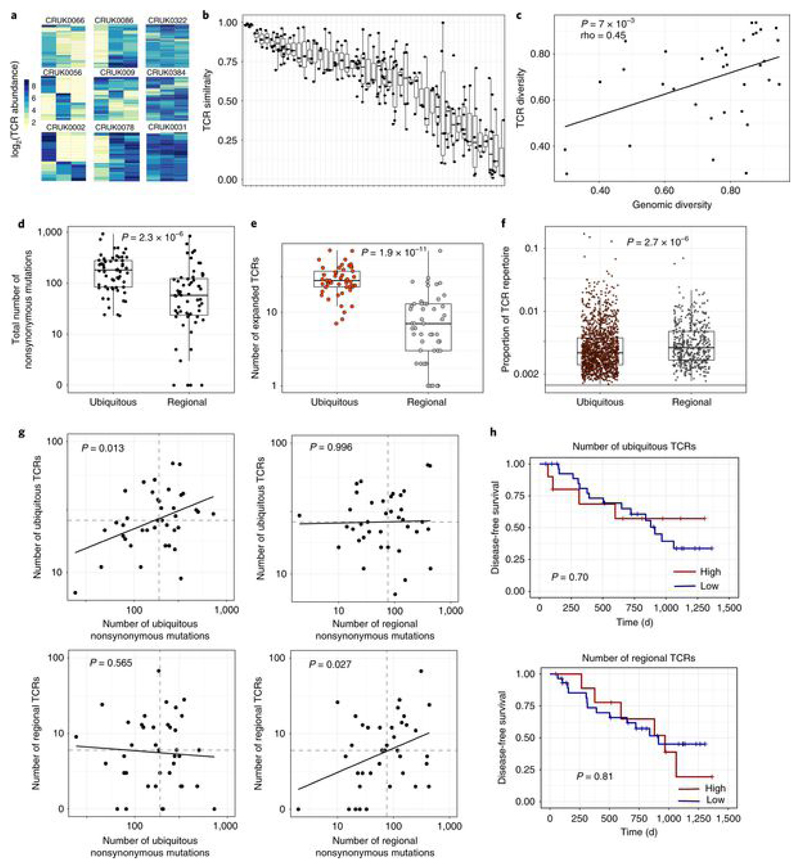
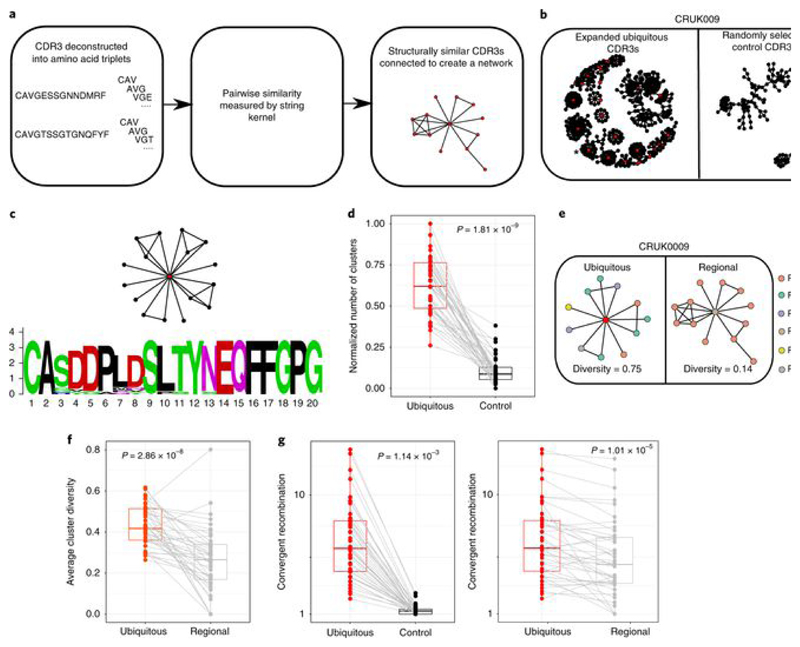
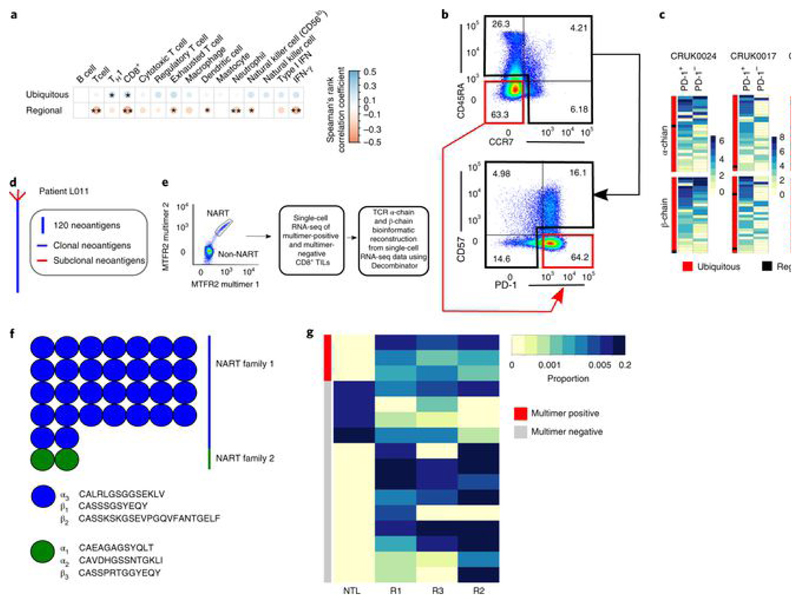
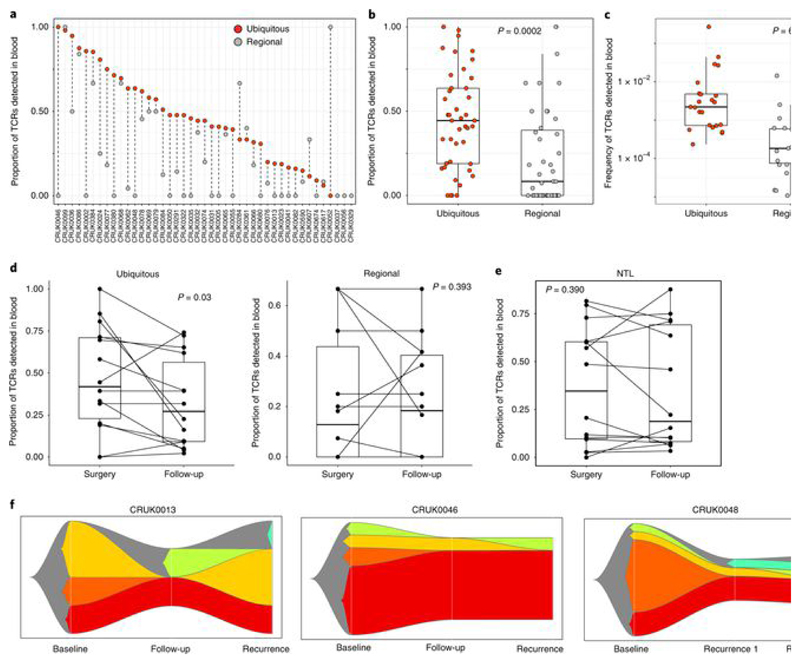
Somatic mutations together with immunoediting drive extensive heterogeneity within non-small-cell lung cancer (NSCLC). Herein we examine heterogeneity of the T cell antigen receptor (TCR) repertoire. The number of TCR sequences selectively expanded in tumors varies within and between tumors and correlates with the number of nonsynonymous mutations. Expanded TCRs can be subdivided into TCRs found in all tumor regions (ubiquitous) and those present in a subset of regions (regional). The number of ubiquitous and regional TCRs correlates with the number of ubiquitous and regional nonsynonymous mutations, respectively. Expanded TCRs form part of clusters of TCRs of similar sequence, suggestive of a spatially constrained antigen-driven process. CD8+ tumor-infiltrating lymphocytes harboring ubiquitous TCRs display a dysfunctional tissue-resident phenotype. Ubiquitous TCRs are preferentially detected in the blood at the time of tumor resection as compared to routine follow-up. These findings highlight a noninvasive method to identify and track relevant tumor-reactive TCRs for use in adoptive T cell immunotherapy.
Conflict of interest statement
Figures

Comment in
-
Tracing evolution reveals new biomarkers.Nat Rev Clin Oncol. 2020 Jan;17(1):5. doi: 10.1038/s41571-019-0295-0. Nat Rev Clin Oncol. 2020. PMID: 31645685 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
- FC001169/MRC_/Medical Research Council/United Kingdom
- 20265/CRUK_/Cancer Research UK/United Kingdom
- FC001169/CRUK_/Cancer Research UK/United Kingdom
- 20764/CRUK_/Cancer Research UK/United Kingdom
- 24314/CRUK_/Cancer Research UK/United Kingdom
- 16463/CRUK_/Cancer Research UK/United Kingdom
- 19310/CRUK_/Cancer Research UK/United Kingdom
- 28990/CRUK_/Cancer Research UK/United Kingdom
- 22246/CRUK_/Cancer Research UK/United Kingdom
- FC001169/ARC_/Arthritis Research UK/United Kingdom
- 17786/CRUK_/Cancer Research UK/United Kingdom
- A17786/CRUK_/Cancer Research UK/United Kingdom
- 30025/CRUK_/Cancer Research UK/United Kingdom
- A22246/CRUK_/Cancer Research UK/United Kingdom
- MC_UP_1203/1/MRC_/Medical Research Council/United Kingdom
- 21999/CRUK_/Cancer Research UK/United Kingdom
- MR/M009033/1/MRC_/Medical Research Council/United Kingdom
- 25253/CRUK_/Cancer Research UK/United Kingdom
- 24956/CRUK_/Cancer Research UK/United Kingdom
- MR/N000838/1/MRC_/Medical Research Council/United Kingdom
- 211179/Z/18/Z/WT_/Wellcome Trust/United Kingdom
- A20764/CRUK_/Cancer Research UK/United Kingdom
- 20466/CRUK_/Cancer Research UK/United Kingdom
- FC001169/WT_/Wellcome Trust/United Kingdom
- WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
