

Cycloaddition Strategies for the Synthesis of Diverse Heterocyclic Spirocycles for Fragment-Based Drug Discovery
- PMID: 31598091
- PMCID: PMC6774287
- DOI: 10.1002/ejoc.201900847
Cycloaddition Strategies for the Synthesis of Diverse Heterocyclic Spirocycles for Fragment-Based Drug Discovery
Abstract
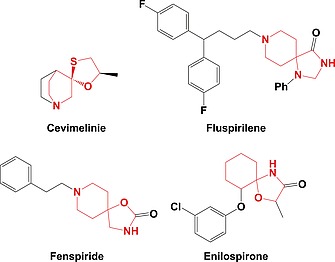
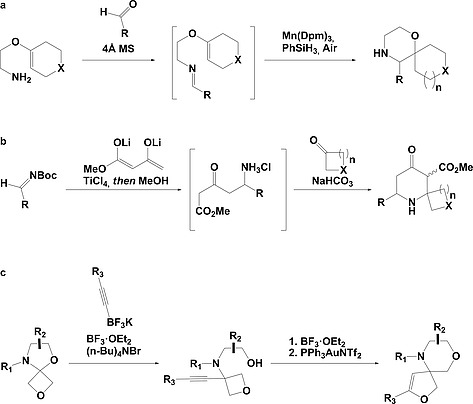
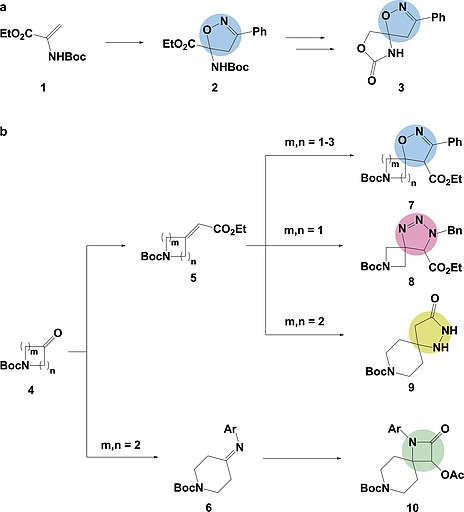
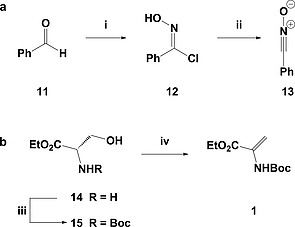
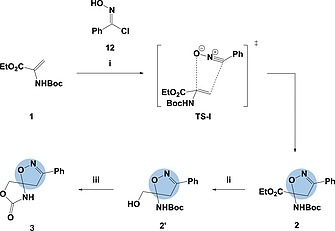
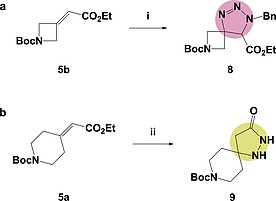
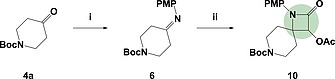
In recent years the pharmaceutical industry has benefited from the advances made in fragment-based drug discovery (FBDD) with more than 30 fragment-derived drugs currently marketed or progressing through clinical trials. The success of fragment-based drug discovery is entirely dependent upon the composition of the fragment screening libraries used. Heterocycles are prevalent within marketed drugs due to the role they play in providing binding interactions; consequently, heterocyclic fragments are important components of FBDD libraries. Current screening libraries are dominated by flat, sp2-rich compounds, primarily owing to their synthetic tractability, despite the superior physicochemical properties displayed by more three-dimensional scaffolds. Herein, we report step-efficient routes to a number of biologically relevant, fragment-like heterocyclic spirocycles. The use of both electron-deficient and electron-rich 2-atom donors was explored in complexity-generating [3+2]-cycloadditions to furnish products in 3 steps from commercially available starting materials. The resulting compounds were primed for further fragment elaboration through the inclusion of synthetic handles from the outset of the syntheses.
Keywords: Cycloaddition; Diversity‐oriented synthesis; Fragment‐based drug discovery; Heterocycles; Spirocycles.
© 2019 The Authors. Published by Wiley‐VCH Verlag GmbH & Co. KGaA.
Figures
References
-
- Ertl P., Jelfs S., Mühlbacher J., Schuffenhauer A. and Selzer P., J. Med. Chem., 2006, 49, 4568–4573. - PubMed
-
- Pitt W. R., Parry D. M., Perry B. G. and Groom C. R., J. Med. Chem., 2009, 52, 2952–2963. - PubMed
-
- Macarron R., Drug Discovery Today, 2006, 11, 277–279. - PubMed
-
- Macarron R., Banks M. N., Bojanic D., Burns D. J., Cirovic D. A., Garyantes T., Green D. V. S., Hertzberg R. P., Janzen W. P., Paslay J. W., et al., Nature, 2011, 10, 188–195. - PubMed
-
- Chan P. F., Holmes D. J. and Payne D. J., Drug Discov. Today Ther. Strateg., 2004, 1, 519–527.
LinkOut - more resources
Full Text Sources
Molecular Biology Databases