

Transcriptional regulatory model of fibrosis progression in the human lung
- PMID: 31600171
- PMCID: PMC6948862
- DOI: 10.1172/jci.insight.131597
Transcriptional regulatory model of fibrosis progression in the human lung
Abstract
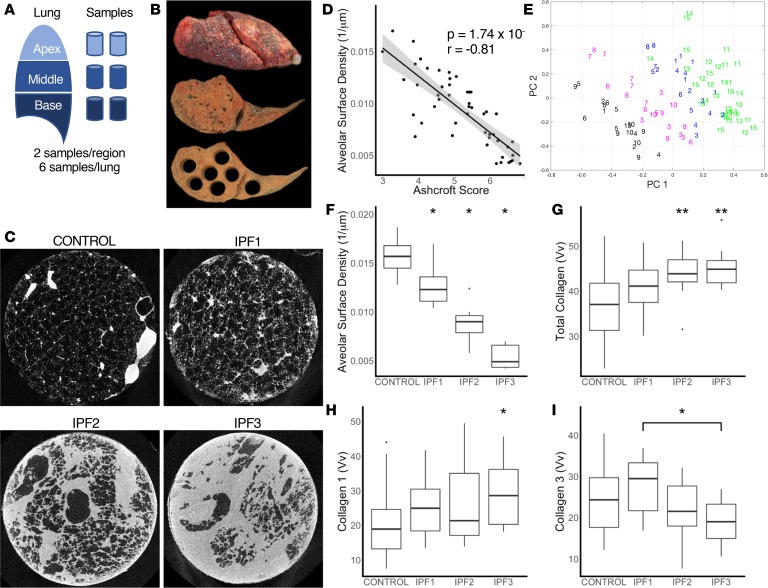
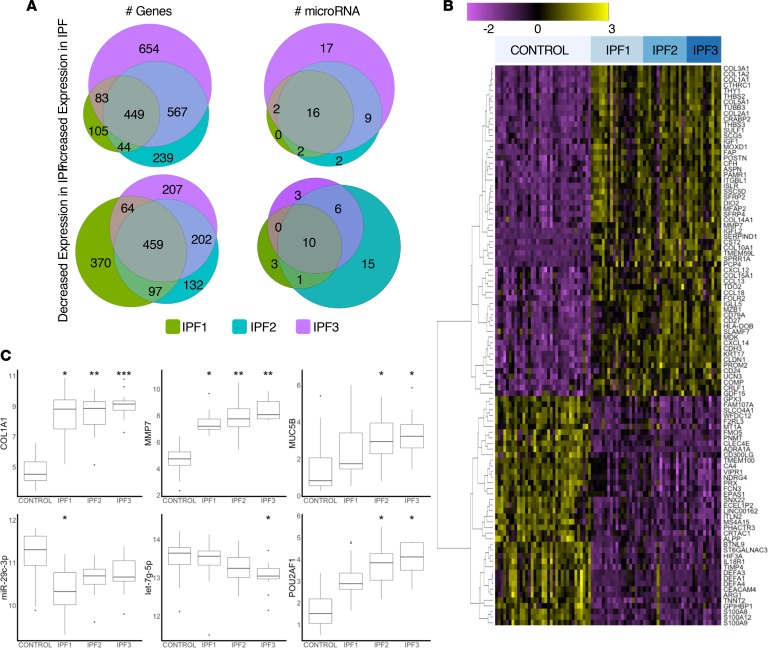
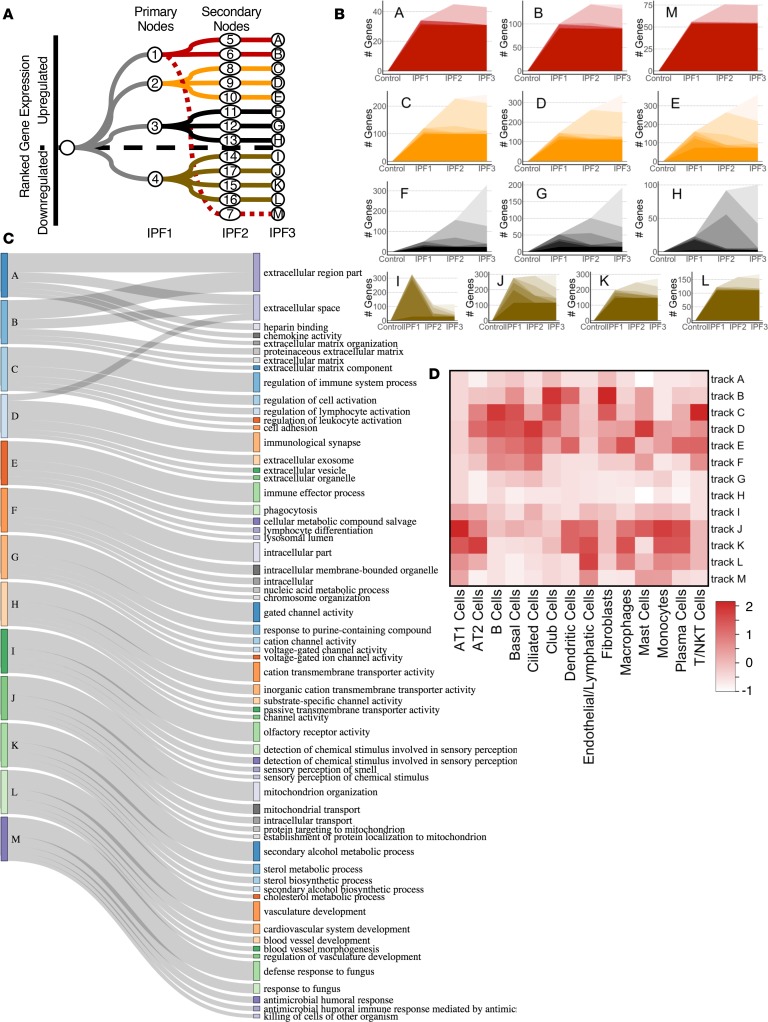
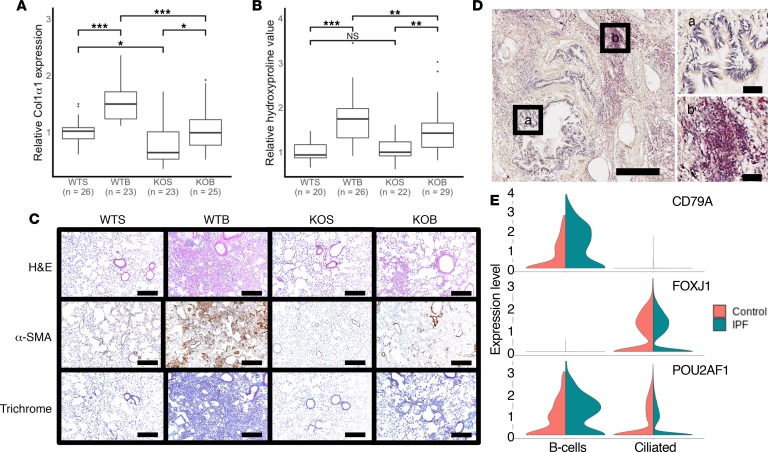
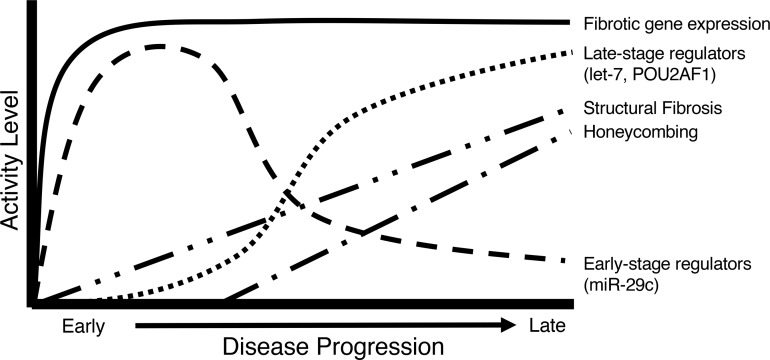
To develop a systems biology model of fibrosis progression within the human lung we performed RNA sequencing and microRNA analysis on 95 samples obtained from 10 idiopathic pulmonary fibrosis (IPF) and 6 control lungs. Extent of fibrosis in each sample was assessed by microCT-measured alveolar surface density (ASD) and confirmed by histology. Regulatory gene expression networks were identified using linear mixed-effect models and dynamic regulatory events miner (DREM). Differential gene expression analysis identified a core set of genes increased or decreased before fibrosis was histologically evident that continued to change with advanced fibrosis. DREM generated a systems biology model (www.sb.cs.cmu.edu/IPFReg) that identified progressively divergent gene expression tracks with microRNAs and transcription factors that specifically regulate mild or advanced fibrosis. We confirmed model predictions by demonstrating that expression of POU2AF1, previously unassociated with lung fibrosis but proposed by the model as regulator, is increased in B lymphocytes in IPF lungs and that POU2AF1-knockout mice were protected from bleomycin-induced lung fibrosis. Our results reveal distinct regulation of gene expression changes in IPF tissue that remained structurally normal compared with moderate or advanced fibrosis and suggest distinct regulatory mechanisms for each stage.
Keywords: Fibrosis; Pulmonology.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
