

Hi-C guided assemblies reveal conserved regulatory topologies on X and autosomes despite extensive genome shuffling
- PMID: 31601616
- PMCID: PMC6824461
- DOI: 10.1101/gad.328971.119
Hi-C guided assemblies reveal conserved regulatory topologies on X and autosomes despite extensive genome shuffling
Abstract
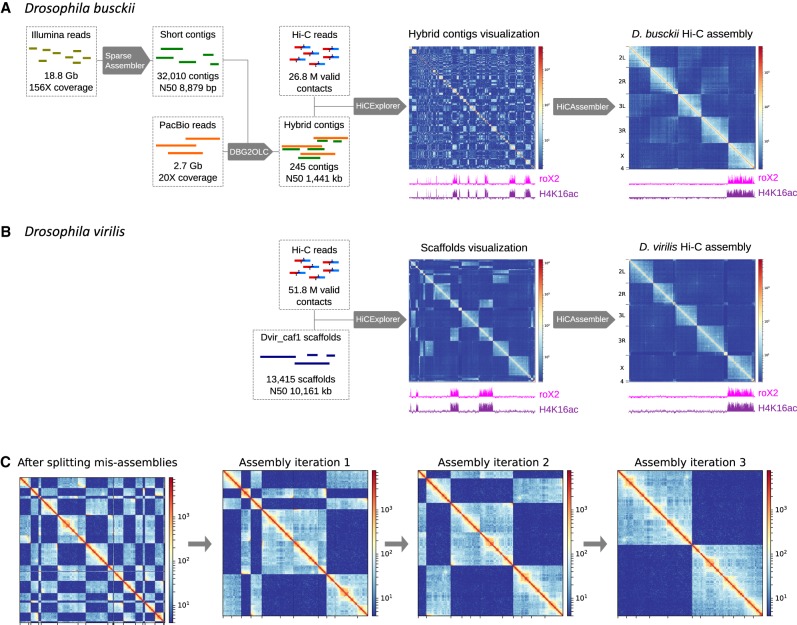
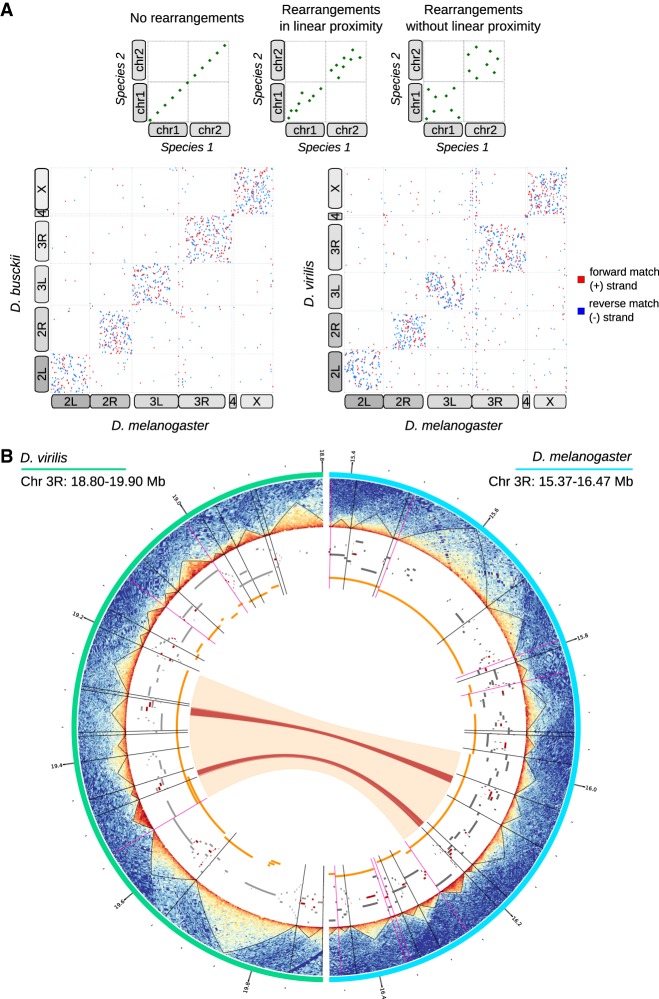
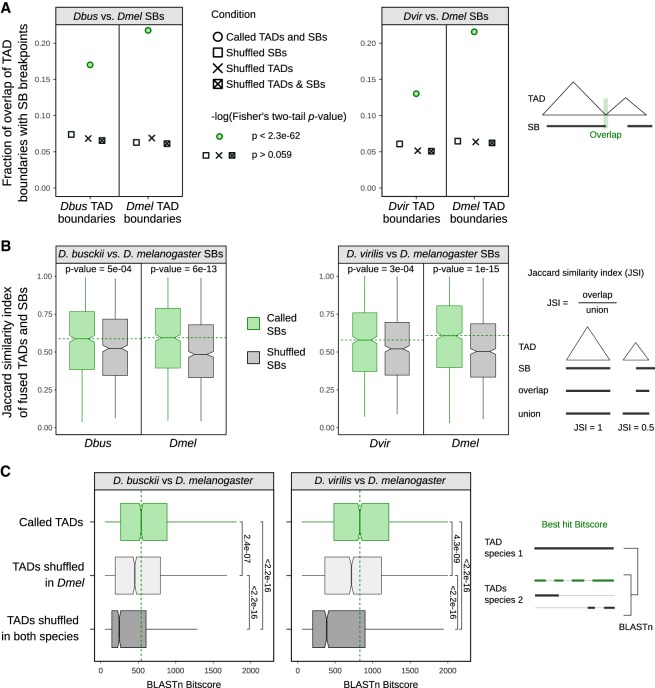
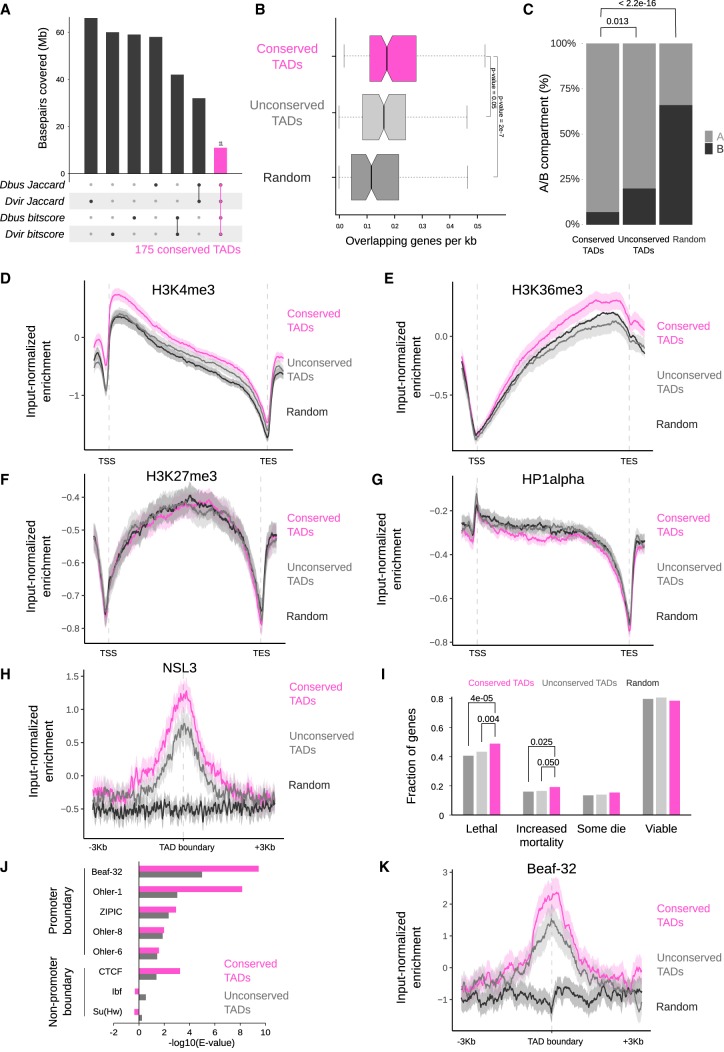
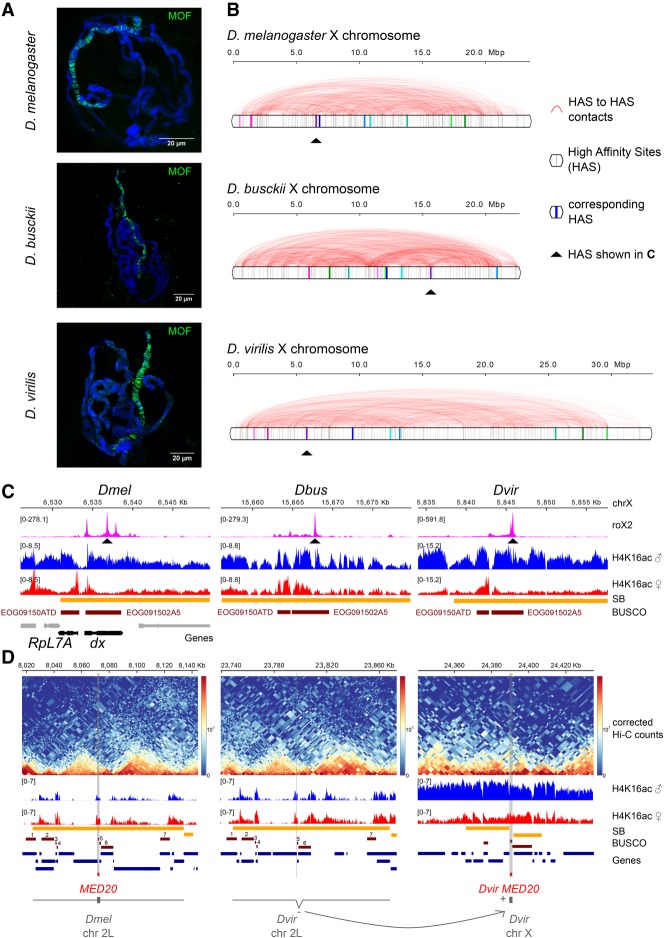
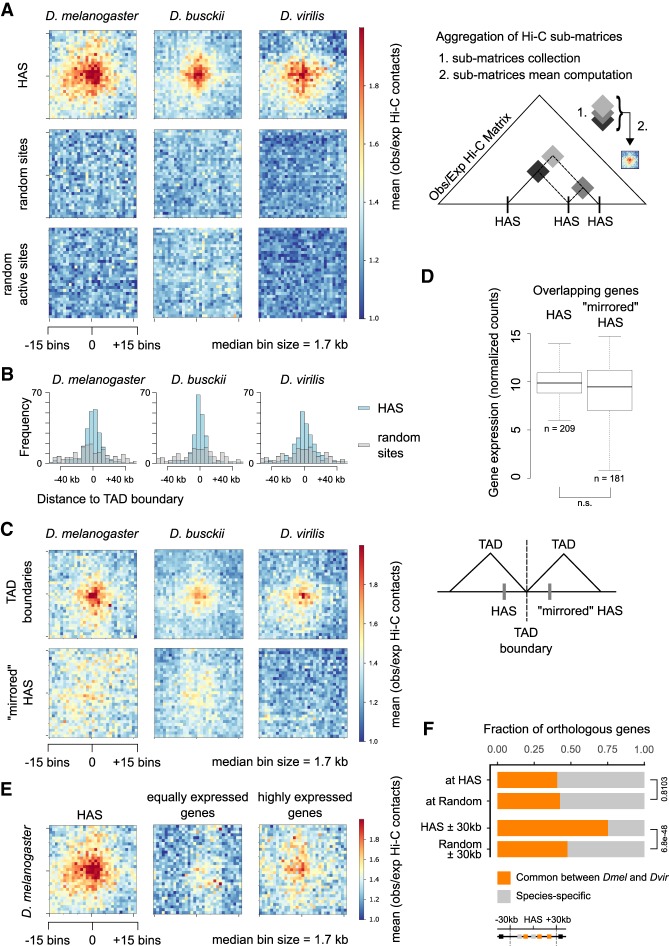
Genome rearrangements that occur during evolution impose major challenges on regulatory mechanisms that rely on three-dimensional genome architecture. Here, we developed a scaffolding algorithm and generated chromosome-length assemblies from Hi-C data for studying genome topology in three distantly related Drosophila species. We observe extensive genome shuffling between these species with one synteny breakpoint after approximately every six genes. A/B compartments, a set of large gene-dense topologically associating domains (TADs), and spatial contacts between high-affinity sites (HAS) located on the X chromosome are maintained over 40 million years, indicating architectural conservation at various hierarchies. Evolutionary conserved genes cluster in the vicinity of HAS, while HAS locations appear evolutionarily flexible, thus uncoupling functional requirement of dosage compensation from individual positions on the linear X chromosome. Therefore, 3D architecture is preserved even in scenarios of thousands of rearrangements highlighting its relevance for essential processes such as dosage compensation of the X chromosome.
Keywords: HiC; X chromosome; chromosome topology; dosage compensation.
© 2019 Renschler et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases