

Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility
- PMID: 31604244
- PMCID: PMC7241648
- DOI: 10.1126/science.aav7188
Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility
Abstract
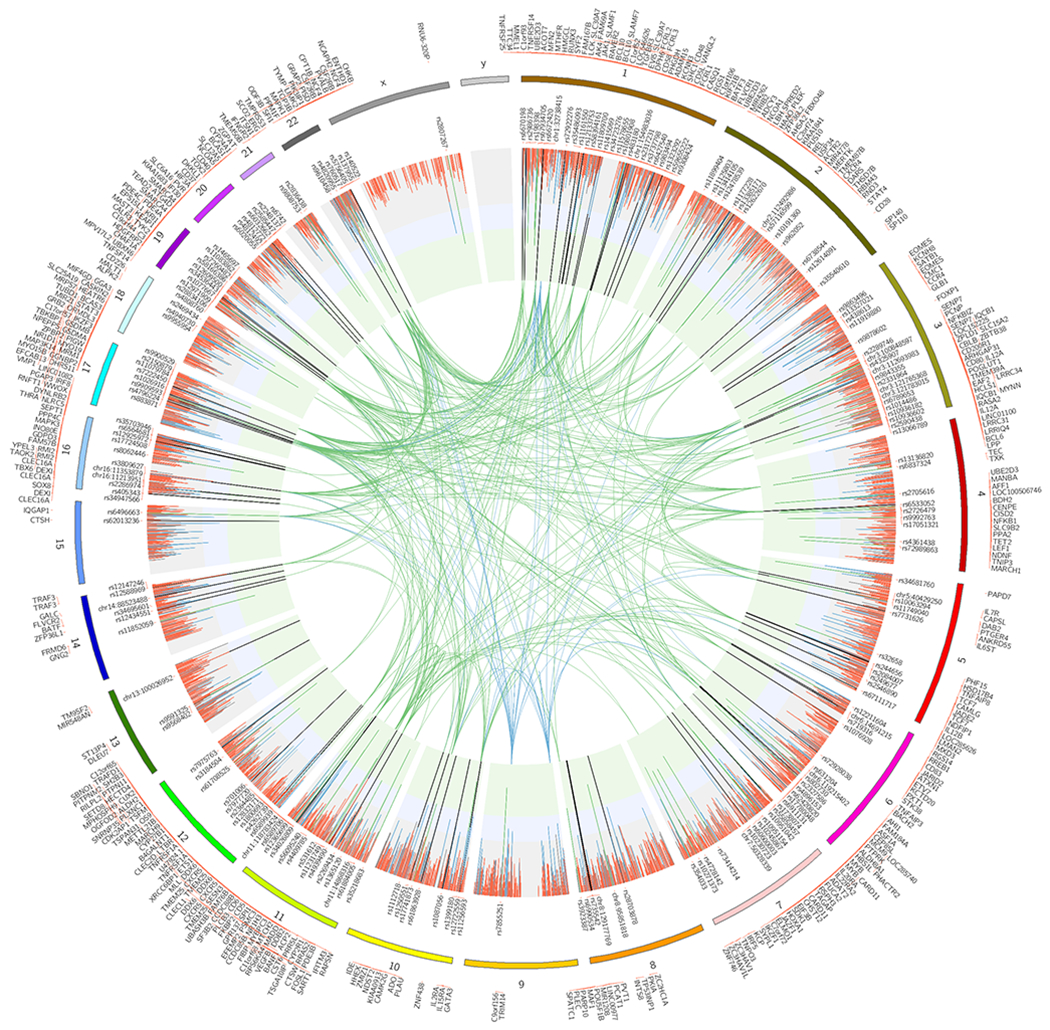
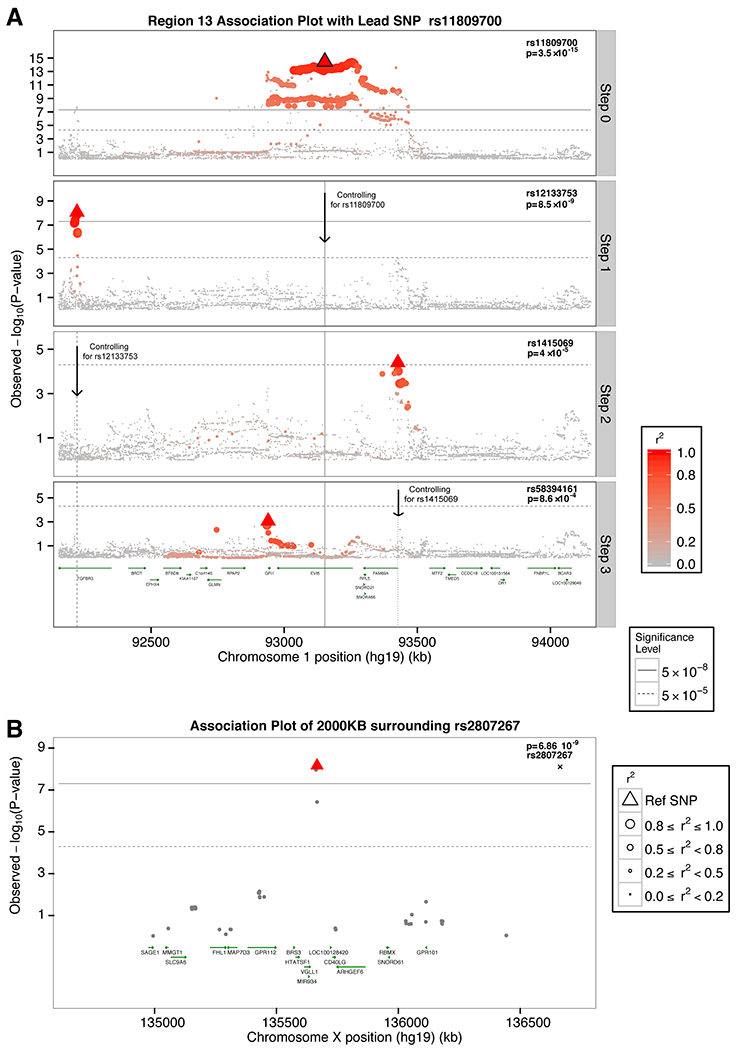
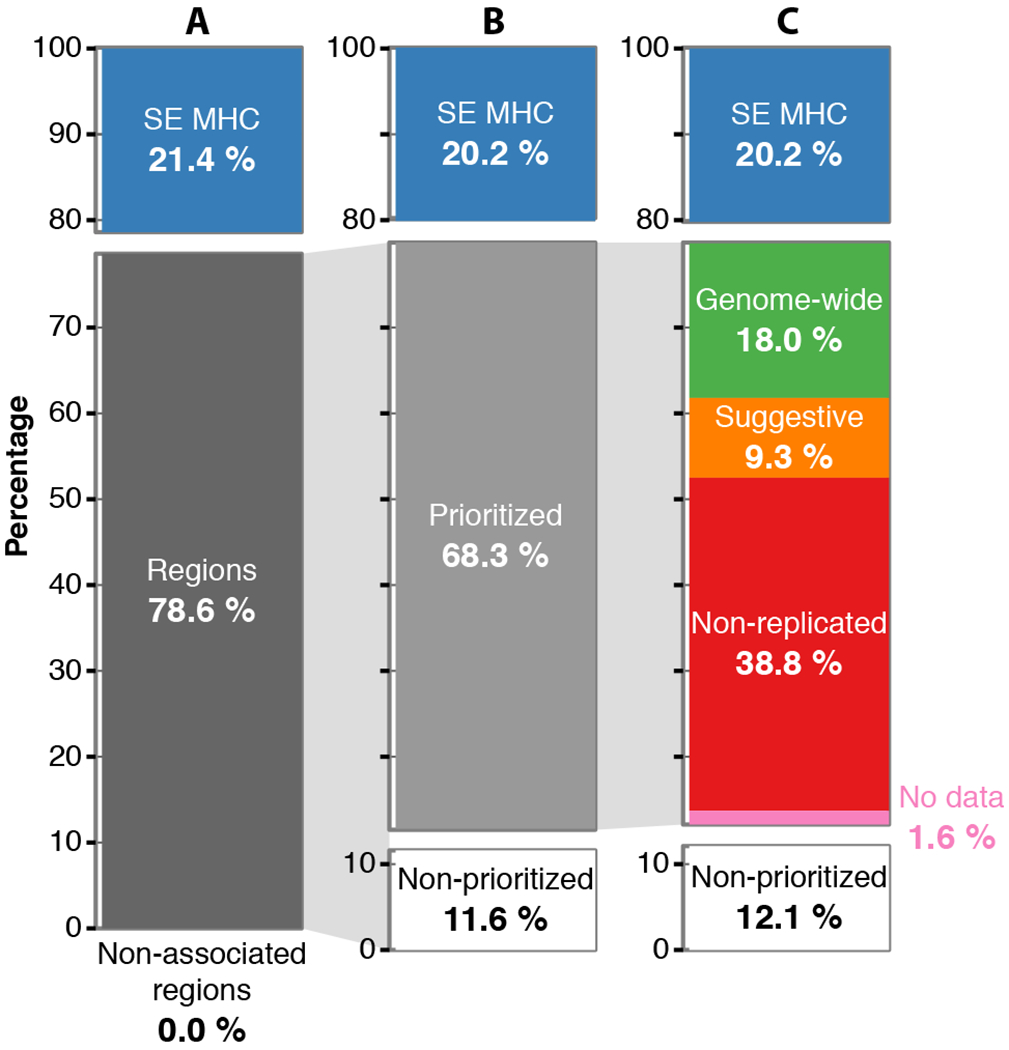
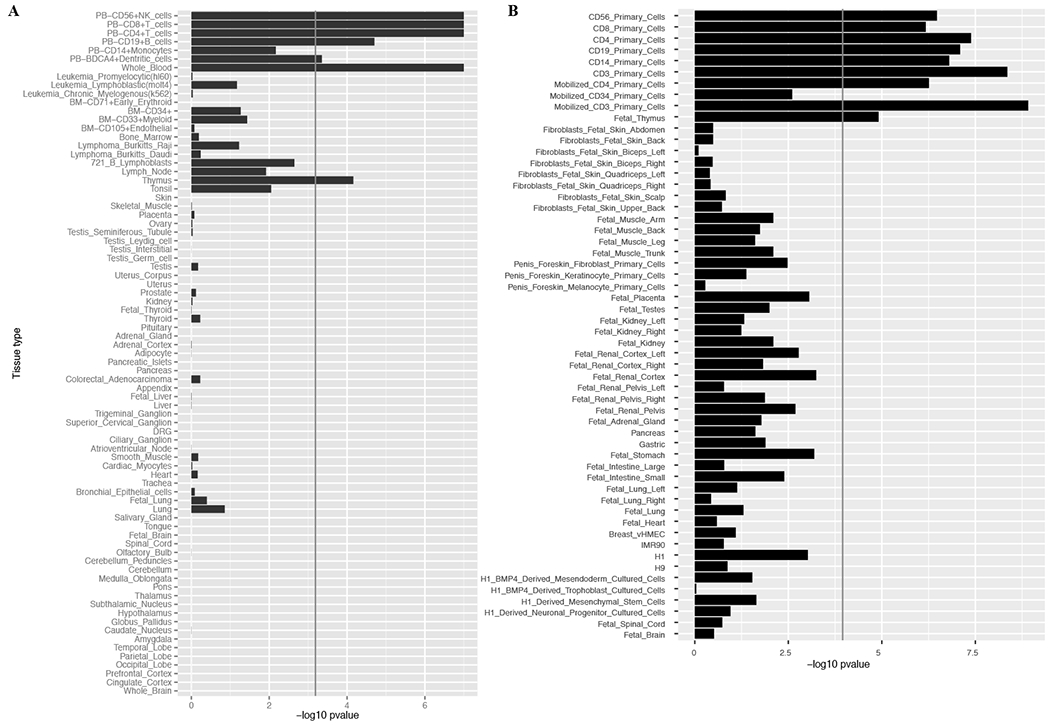
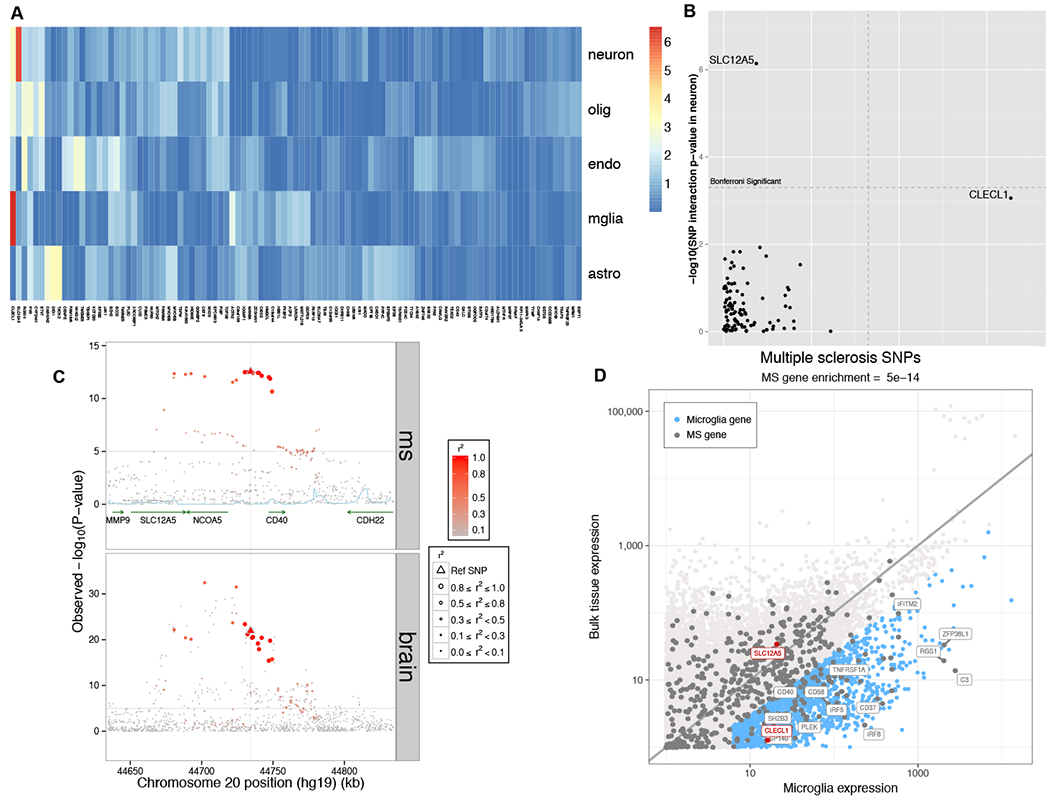
We analyzed genetic data of 47,429 multiple sclerosis (MS) and 68,374 control subjects and established a reference map of the genetic architecture of MS that includes 200 autosomal susceptibility variants outside the major histocompatibility complex (MHC), one chromosome X variant, and 32 variants within the extended MHC. We used an ensemble of methods to prioritize 551 putative susceptibility genes that implicate multiple innate and adaptive pathways distributed across the cellular components of the immune system. Using expression profiles from purified human microglia, we observed enrichment for MS genes in these brain-resident immune cells, suggesting that these may have a role in targeting an autoimmune process to the central nervous system, although MS is most likely initially triggered by perturbation of peripheral immune responses.
Copyright © 2019, American Association for the Advancement of Science.
Conflict of interest statement
Figures

Comment in
-
Unraveling susceptibility to multiple sclerosis.Science. 2019 Sep 27;365(6460):1383-1384. doi: 10.1126/science.aay1439. Science. 2019. PMID: 31604260 No abstract available.
-
Putting multiple sclerosis on the genomic map.Nat Rev Neurol. 2019 Dec;15(12):686-687. doi: 10.1038/s41582-019-0279-9. Nat Rev Neurol. 2019. PMID: 31619774 No abstract available.
References
-
- International Multiple Sclerosis Genetics Consortium et al., Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med 357, 851–862 (2007). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
