

MetaCell: analysis of single-cell RNA-seq data using K-nn graph partitions
- PMID: 31604482
- PMCID: PMC6790056
- DOI: 10.1186/s13059-019-1812-2
MetaCell: analysis of single-cell RNA-seq data using K-nn graph partitions
Abstract
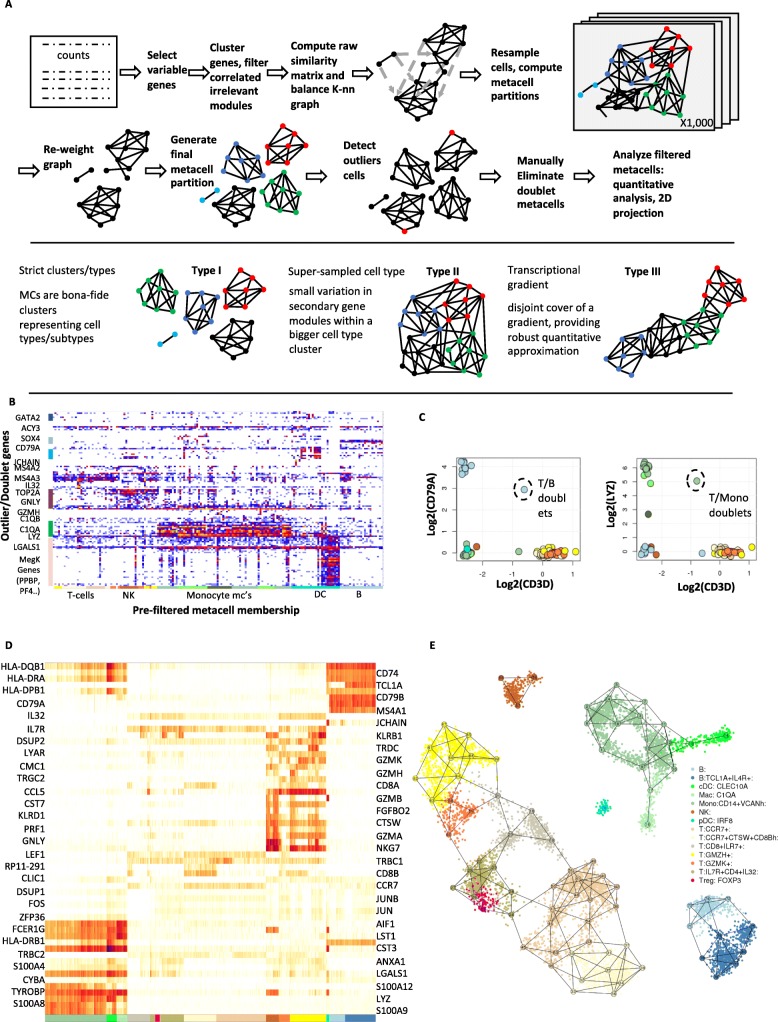
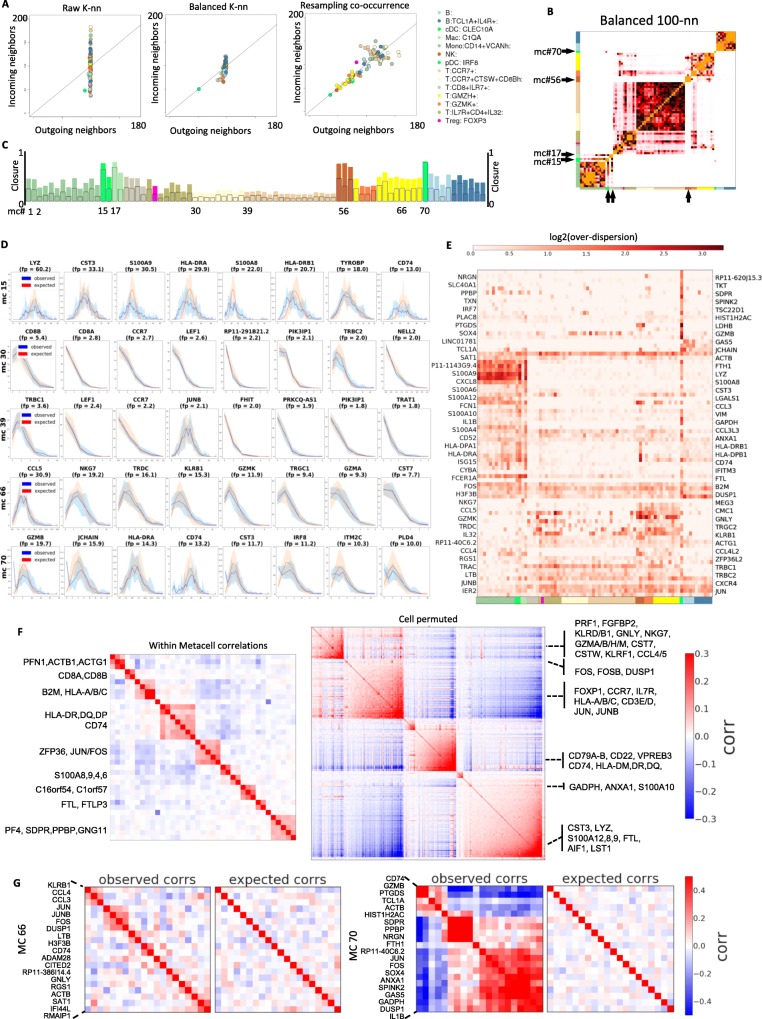
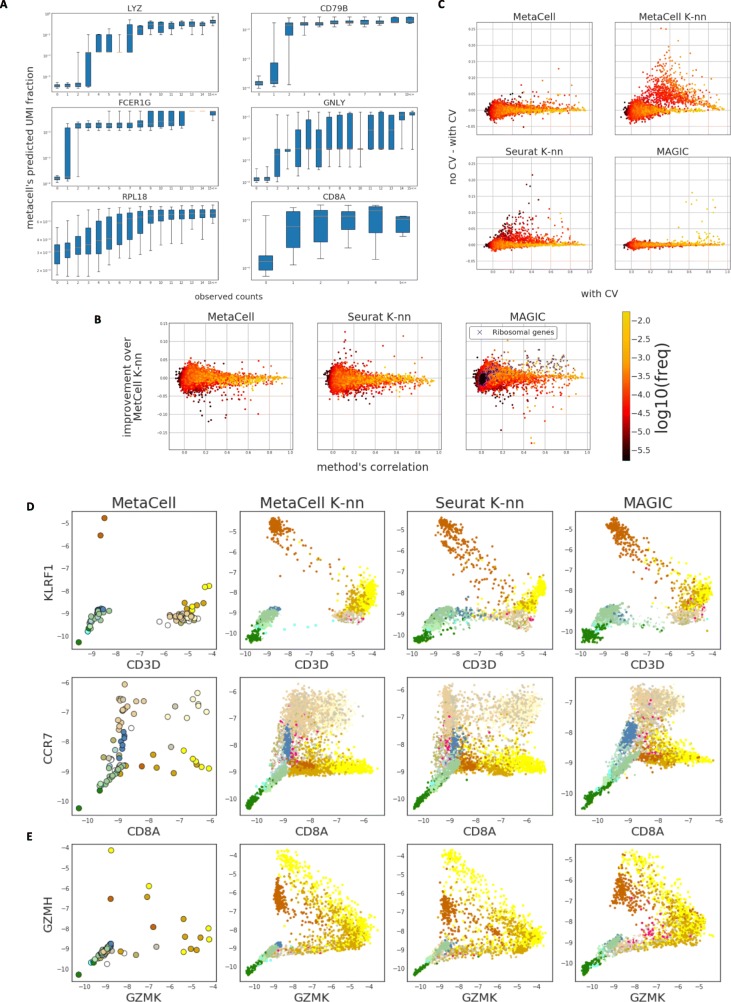
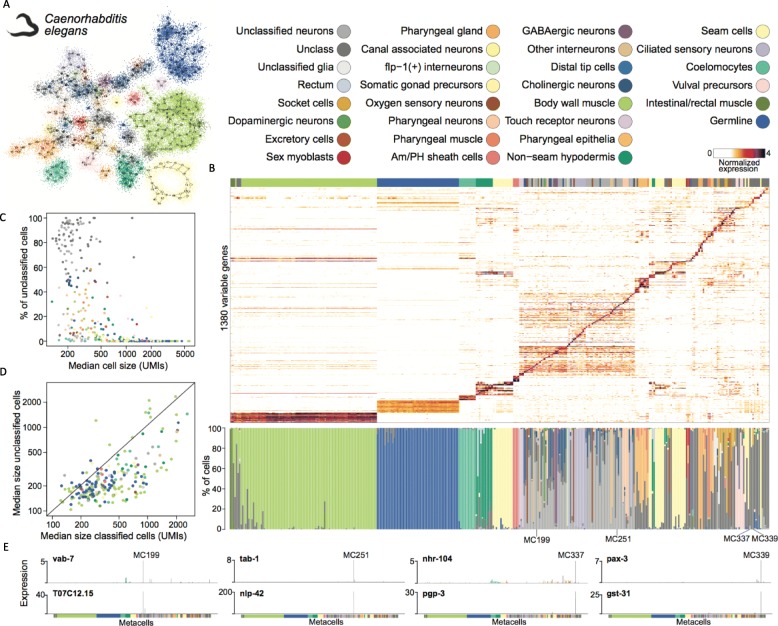
scRNA-seq profiles each represent a highly partial sample of mRNA molecules from a unique cell that can never be resampled, and robust analysis must separate the sampling effect from biological variance. We describe a methodology for partitioning scRNA-seq datasets into metacells: disjoint and homogenous groups of profiles that could have been resampled from the same cell. Unlike clustering analysis, our algorithm specializes at obtaining granular as opposed to maximal groups. We show how to use metacells as building blocks for complex quantitative transcriptional maps while avoiding data smoothing. Our algorithms are implemented in the MetaCell R/C++ software package.
Keywords: Clustering; Graph partition; Multinomial distribution; RNA-seq; Sampling variance; Smoothing; scRNA-seq.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
