

Neoantigen Dissimilarity to the Self-Proteome Predicts Immunogenicity and Response to Immune Checkpoint Blockade
- PMID: 31606370
- PMCID: PMC6813910
- DOI: 10.1016/j.cels.2019.08.009
Neoantigen Dissimilarity to the Self-Proteome Predicts Immunogenicity and Response to Immune Checkpoint Blockade
Abstract
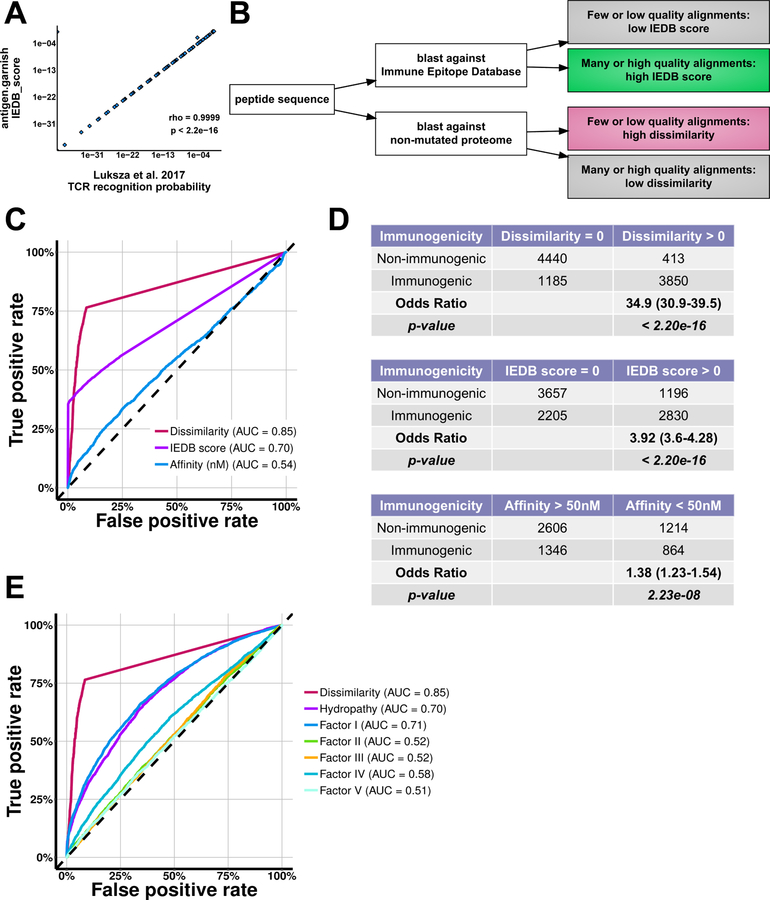
Despite improved methods for MHC affinity prediction, the vast majority of computationally predicted tumor neoantigens are not immunogenic experimentally, indicating that high-quality neoantigens are beyond current algorithms to discern. To enrich for neoantigens with the greatest likelihood of immunogenicity, we developed an analytic method to parse neoantigen quality through rational biological criteria across five clinical datasets for 318 cancer patients. We explored four quality metrics, including analysis of dissimilarity to the non-mutated proteome that was predictive of peptide immunogenicity. In patient tumors, neoantigens with high dissimilarity were unique, enriched for hydrophobic sequences, and correlated with survival after PD-1 checkpoint therapy in patients with non-small cell lung cancer independent of predicted MHC affinity. We incorporated our neoantigen quality analysis methodology into an open-source tool, antigen.garnish, to predict immunogenic peptides from bulk computationally predicted neoantigens for which the immunogenic "hit rate" is currently low.
Keywords: immune checkpoint blockade; immunogenicity prediction; neoantigen; tumor immunology.
Copyright © 2019 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests
L.P.R. and A.J.R. declare no competing interests.
Figures



References
-
- Bhattacharya R, Sivakumar A, Tokheim C, Guthrie VB, Anagnostou V, Velculescu VE, and Karchin R (2017). Evaluation of machine learning methods to predict peptide binding to MHC Class I proteins. BioRxiv 154757.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
