

Diagnostic utility of transcriptome sequencing for rare Mendelian diseases
- PMID: 31607746
- PMCID: PMC7405636
- DOI: 10.1038/s41436-019-0672-1
Diagnostic utility of transcriptome sequencing for rare Mendelian diseases
Abstract
Purpose: We investigated the value of transcriptome sequencing (RNAseq) in ascertaining the consequence of DNA variants on RNA transcripts to improve the diagnostic rate from exome or genome sequencing for undiagnosed Mendelian diseases spanning a wide spectrum of clinical indications.
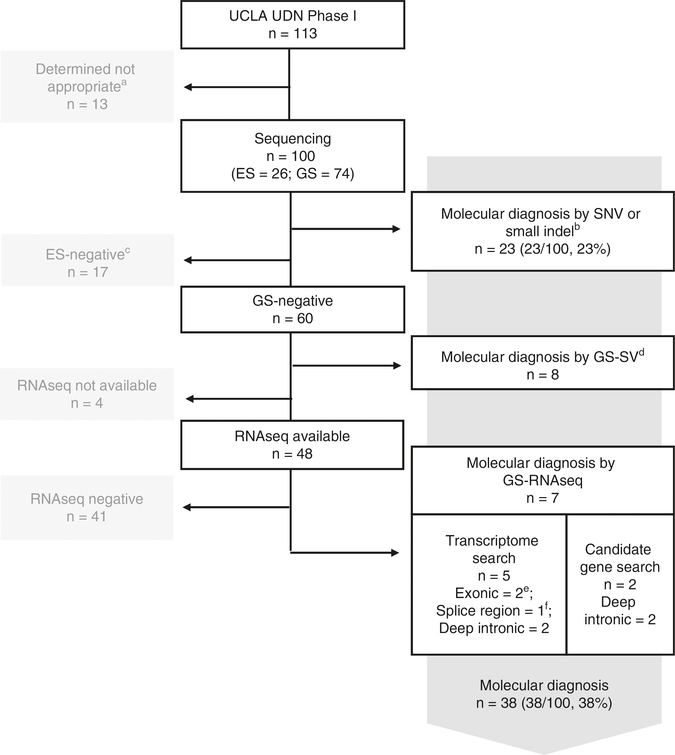
Methods: From 234 subjects referred to the Undiagnosed Diseases Network, University of California-Los Angeles clinical site between July 2014 and August 2018, 113 were enrolled for high likelihood of having rare undiagnosed, suspected genetic conditions despite thorough prior clinical evaluation. Exome or genome sequencing and RNAseq were performed, and RNAseq data was integrated with genome sequencing data for DNA variant interpretation genome-wide.
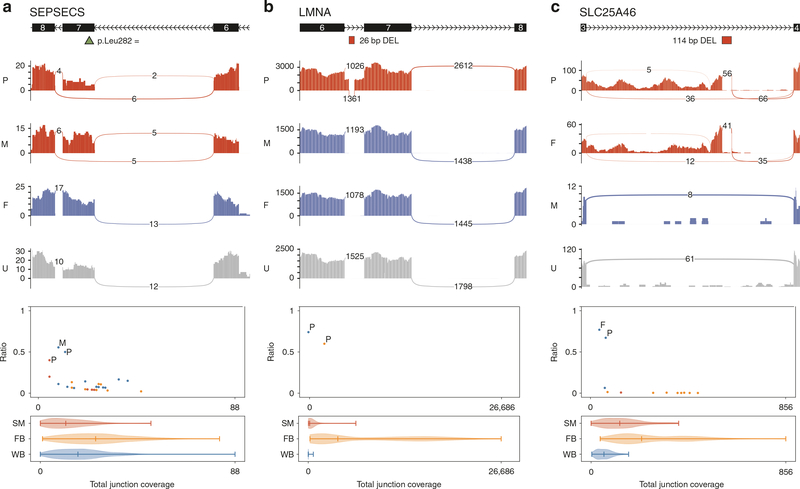
Results: The molecular diagnostic rate by exome or genome sequencing was 31%. Integration of RNAseq with genome sequencing resulted in an additional seven cases with clear diagnosis of a known genetic disease. Thus, the overall molecular diagnostic rate was 38%, and 18% of all genetic diagnoses returned required RNAseq to determine variant causality.
Conclusion: In this rare disease cohort with a wide spectrum of undiagnosed, suspected genetic conditions, RNAseq analysis increased the molecular diagnostic rate above that possible with genome sequencing analysis alone even without availability of the most appropriate tissue type to assess.
Keywords: exome sequencing; genome sequencing; molecular diagnosis; transcriptome sequencing; undiagnosed rare Mendelian diseases.
Conflict of interest statement
DISCLOSURE
The authors declare no conflicts of interest.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
