

Acute Respiratory Barrier Disruption by Ozone Exposure in Mice
- PMID: 31608051
- PMCID: PMC6758598
- DOI: 10.3389/fimmu.2019.02169
Acute Respiratory Barrier Disruption by Ozone Exposure in Mice
Abstract
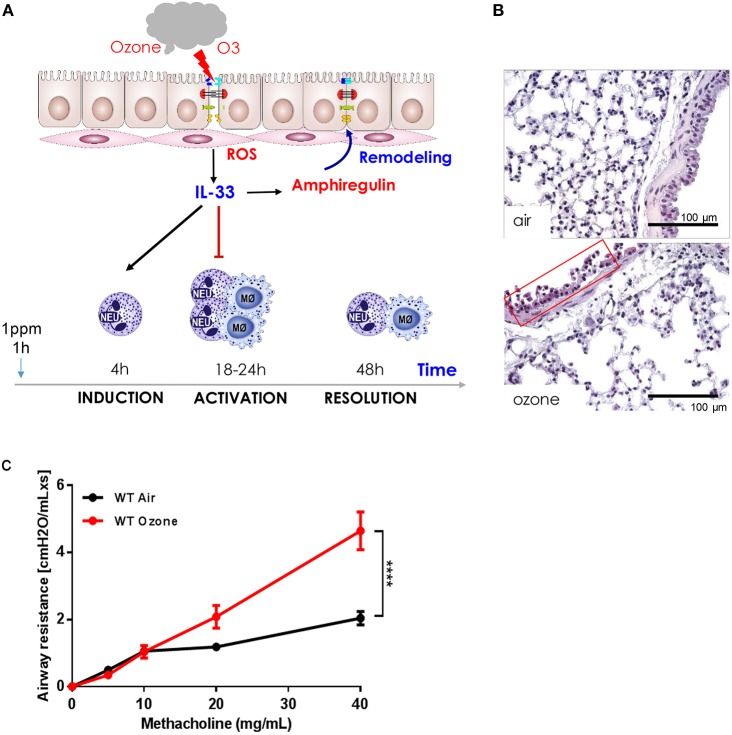
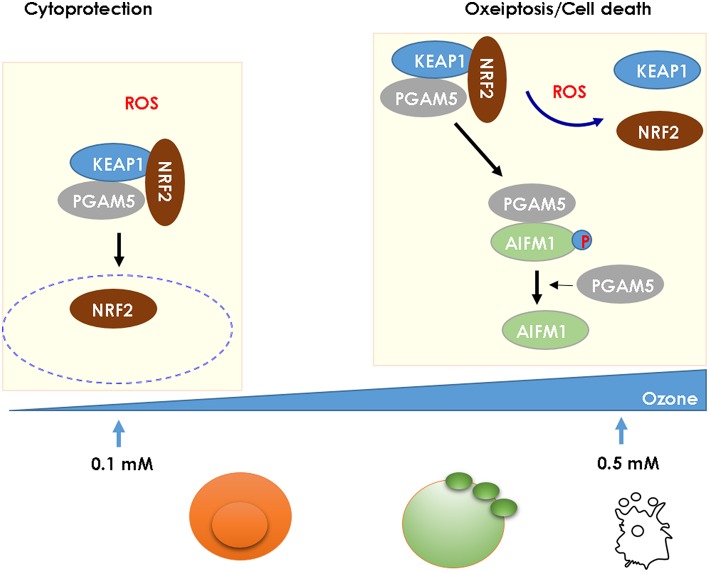
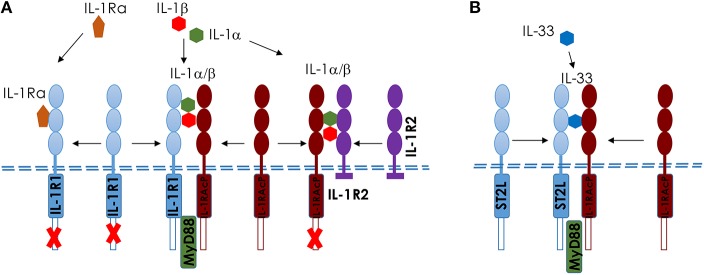
Ozone exposure causes irritation, airway hyperreactivity (AHR), inflammation of the airways, and destruction of alveoli (emphysema), the gas exchange area of the lung in human and mice. This review focuses on the acute disruption of the respiratory epithelial barrier in mice. A single high dose ozone exposure (1 ppm for 1 h) causes first a break of the bronchiolar epithelium within 2 h with leak of serum proteins in the broncho-alveolar space, disruption of epithelial tight junctions and cell death, which is followed at 6 h by ROS activation, AHR, myeloid cell recruitment, and remodeling. High ROS levels activate a novel PGAM5 phosphatase dependent cell-death pathway, called oxeiptosis. Bronchiolar cell wall damage and inflammation upon a single ozone exposure are reversible. However, chronic ozone exposure leads to progressive and irreversible loss of alveolar epithelial cells and alveoli with reduced gas exchange space known as emphysema. It is further associated with chronic inflammation and fibrosis of the lung, resembling other environmental pollutants and cigarette smoke in pathogenesis of asthma, and chronic obstructive pulmonary disease (COPD). Here, we review recent data on the mechanisms of ozone induced injury on the different cell types and pathways with a focus on the role of the IL-1 family cytokines and the related IL-33. The relation of chronic ozone exposure induced lung disease with asthma and COPD and the fact that ozone exacerbates asthma and COPD is emphasized.
Keywords: cell death; inflammation; innate immunity; interleukins; mucus; tight junctions.
Copyright © 2019 Sokolowska, Quesniaux, Akdis, Chung, Ryffel and Togbe.
Figures
Similar articles
-
Oxidative Stress in Ozone-Induced Chronic Lung Inflammation and Emphysema: A Facet of Chronic Obstructive Pulmonary Disease.Front Immunol. 2020 Sep 2;11:1957. doi: 10.3389/fimmu.2020.01957. eCollection 2020. Front Immunol. 2020. PMID: 32983127 Free PMC article. Review.
-
Functional and morphological differences of the lung upon acute and chronic ozone exposure in mice.Sci Rep. 2018 Jul 13;8(1):10611. doi: 10.1038/s41598-018-28261-9. Sci Rep. 2018. PMID: 30006538 Free PMC article.
-
Klotho expression is reduced in COPD airway epithelial cells: effects on inflammation and oxidant injury.Clin Sci (Lond). 2015 Dec;129(12):1011-23. doi: 10.1042/CS20150273. Epub 2015 Jul 10. Clin Sci (Lond). 2015. PMID: 26201096 Free PMC article.
-
Ozone exposure induces respiratory barrier biphasic injury and inflammation controlled by IL-33.J Allergy Clin Immunol. 2018 Sep;142(3):942-958. doi: 10.1016/j.jaci.2017.11.044. Epub 2018 Jan 10. J Allergy Clin Immunol. 2018. PMID: 29331644
-
Airway Epithelial Barrier Dysfunction in Chronic Obstructive Pulmonary Disease: Role of Cigarette Smoke Exposure.Am J Respir Cell Mol Biol. 2018 Feb;58(2):157-169. doi: 10.1165/rcmb.2017-0200TR. Am J Respir Cell Mol Biol. 2018. PMID: 28933915 Review.
Cited by
-
Oxypeucedanin relieves LPS-induced acute lung injury by inhibiting the inflammation and maintaining the integrity of the lung air-blood barrier.Aging (Albany NY). 2022 Aug 18;14(16):6626-6641. doi: 10.18632/aging.204235. Epub 2022 Aug 18. Aging (Albany NY). 2022. PMID: 35985771 Free PMC article.
-
Synergistic Toxicity of Fine Particulate Matter and Ozone and Their Underlying Mechanisms.Toxics. 2025 Mar 24;13(4):236. doi: 10.3390/toxics13040236. Toxics. 2025. PMID: 40278552 Free PMC article. Review.
-
The External Exposome and Allergies: From the Perspective of the Epithelial Barrier Hypothesis.Front Allergy. 2022 Jul 8;3:887672. doi: 10.3389/falgy.2022.887672. eCollection 2022. Front Allergy. 2022. PMID: 35873598 Free PMC article. Review.
-
β-Endorphin (an endogenous opioid) inhibits inflammation, oxidative stress and apoptosis via Nrf-2 in asthmatic murine model.Sci Rep. 2023 Jul 31;13(1):12414. doi: 10.1038/s41598-023-38366-5. Sci Rep. 2023. PMID: 37524754 Free PMC article.
-
The dual role of PGAM5 in inflammation.Exp Mol Med. 2025 Feb;57(2):298-311. doi: 10.1038/s12276-025-01391-7. Epub 2025 Feb 10. Exp Mol Med. 2025. PMID: 39930129 Free PMC article. Review.
References
-
- Wiegman CH, Michaeloudes C, Haji G, Narang P, Clarke CJ, Russell KE, et al. . Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. (2015) 136:769–80. 10.1016/j.jaci.2015.01.046 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical