

KHDC3L mutation causes recurrent pregnancy loss by inducing genomic instability of human early embryonic cells
- PMID: 31609975
- PMCID: PMC6812846
- DOI: 10.1371/journal.pbio.3000468
KHDC3L mutation causes recurrent pregnancy loss by inducing genomic instability of human early embryonic cells
Abstract
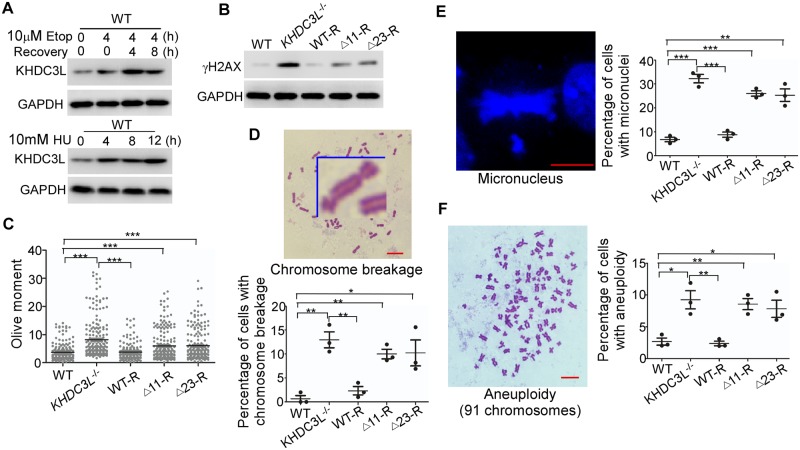
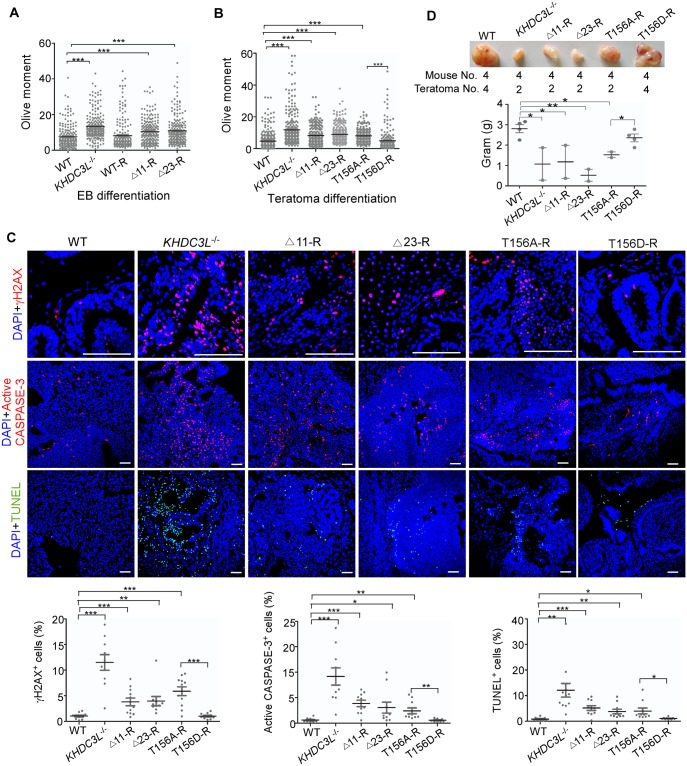
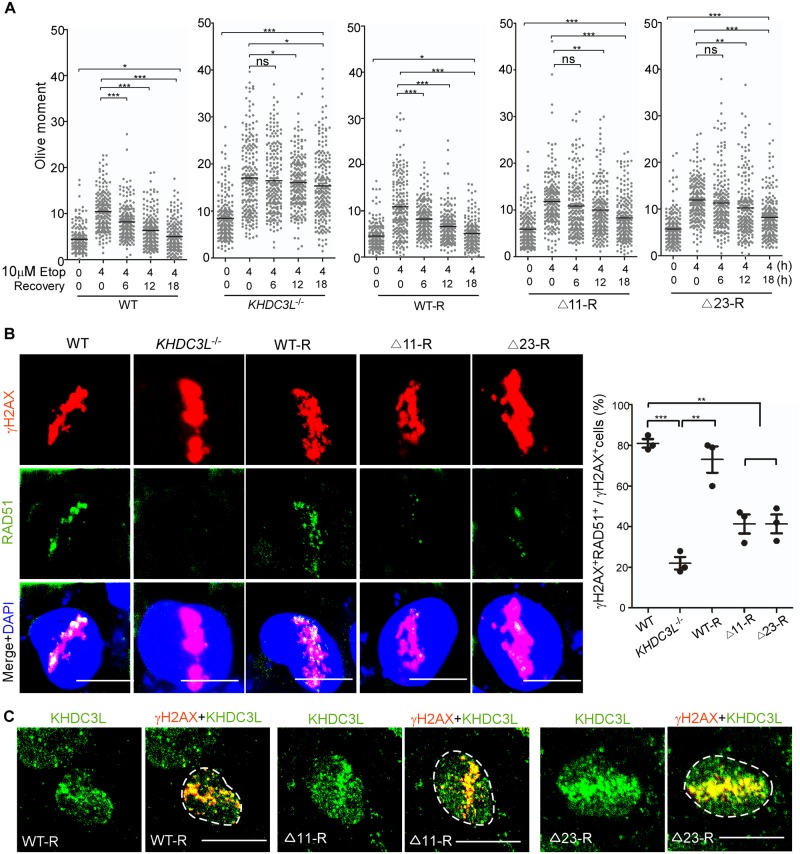
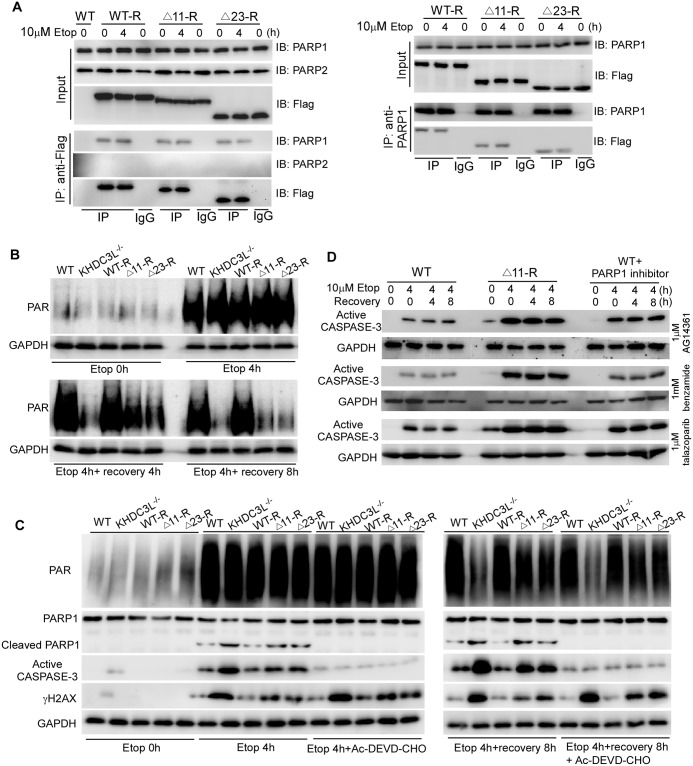
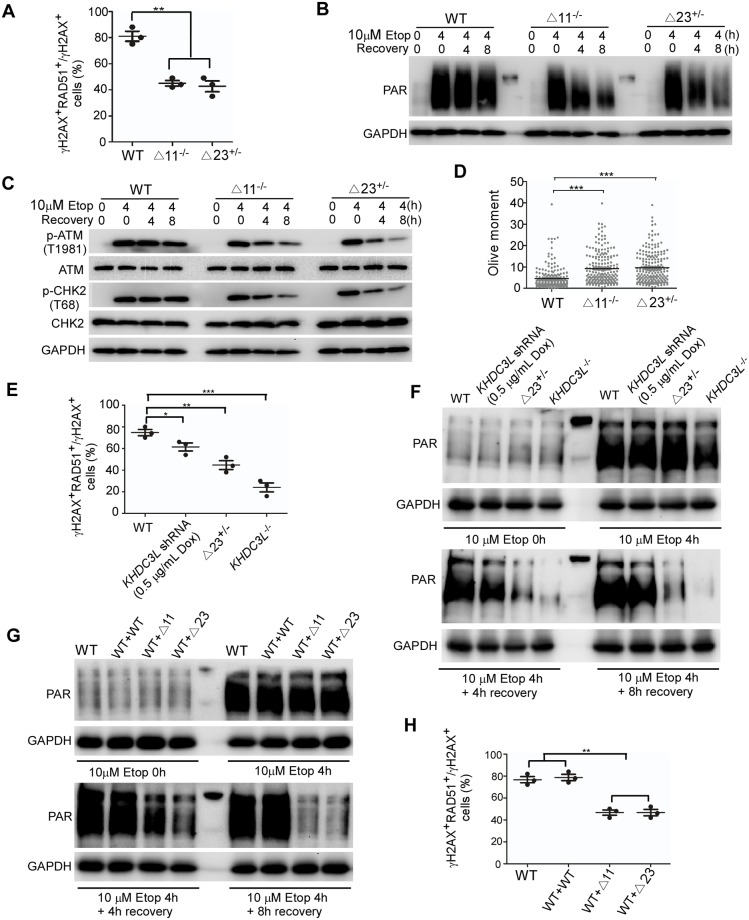
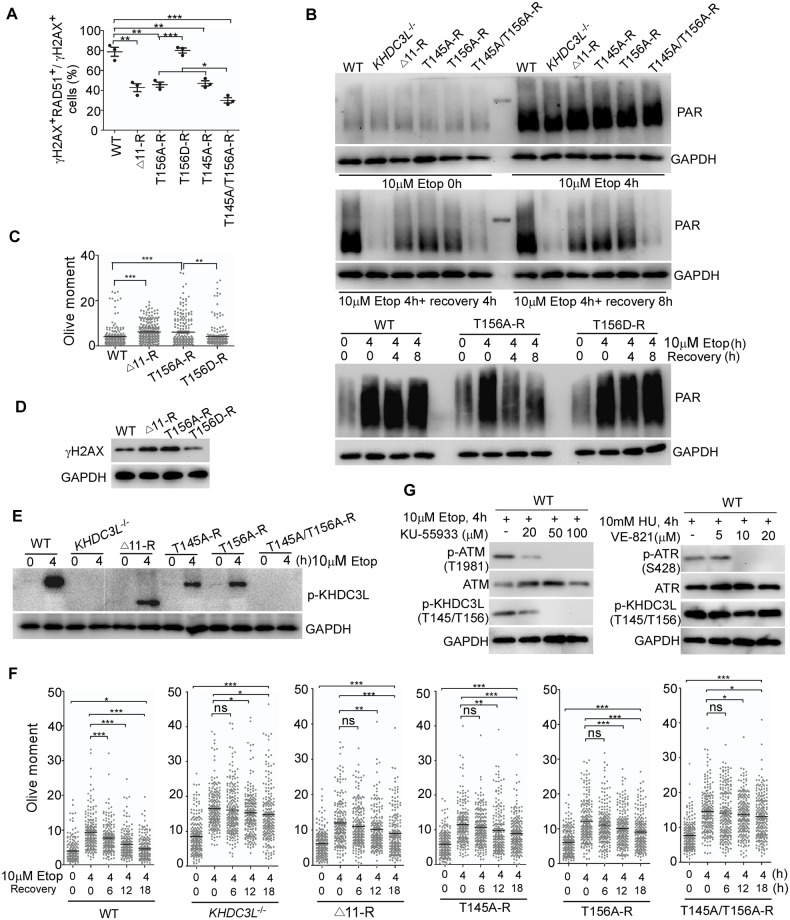
Recurrent pregnancy loss (RPL) is an important complication in reproductive health. About 50% of RPL cases are unexplained, and understanding the genetic basis is essential for its diagnosis and prognosis. Herein, we report causal KH domain containing 3 like (KHDC3L) mutations in RPL. KHDC3L is expressed in human epiblast cells and ensures their genome stability and viability. Mechanistically, KHDC3L binds to poly(ADP-ribose) polymerase 1 (PARP1) to stimulate its activity. In response to DNA damage, KHDC3L also localizes to DNA damage sites and facilitates homologous recombination (HR)-mediated DNA repair. KHDC3L dysfunction causes PARP1 inhibition and HR repair deficiency, which is synthetically lethal. Notably, we identified two critical residues, Thr145 and Thr156, whose phosphorylation by Ataxia-telangiectasia mutated (ATM) is essential for KHDC3L's functions. Importantly, two deletions of KHDC3L (p.E150_V160del and p.E150_V172del) were detected in female RPL patients, both of which harbor a common loss of Thr156 and are impaired in PARP1 activation and HR repair. In summary, our study reveals both KHDC3L as a new RPL risk gene and its critical function in DNA damage repair pathways.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

Similar articles
-
No evidence for mutations in NLRP7, NLRP2 or KHDC3L in women with unexplained recurrent pregnancy loss or infertility.Hum Reprod. 2015 Jan;30(1):232-8. doi: 10.1093/humrep/deu296. Epub 2014 Nov 5. Hum Reprod. 2015. PMID: 25376457 Free PMC article.
-
KH domain containing 3 like (KHDC3L) frame-shift mutation causes both recurrent pregnancy loss and hydatidiform mole.Eur J Obstet Gynecol Reprod Biol. 2021 Apr;259:100-104. doi: 10.1016/j.ejogrb.2021.02.006. Epub 2021 Feb 10. Eur J Obstet Gynecol Reprod Biol. 2021. PMID: 33639414
-
The WD40 domain of FBXW7 is a poly(ADP-ribose)-binding domain that mediates the early DNA damage response.Nucleic Acids Res. 2019 May 7;47(8):4039-4053. doi: 10.1093/nar/gkz058. Nucleic Acids Res. 2019. PMID: 30722038 Free PMC article.
-
DNA repair factors and telomere-chromosome integrity in mammalian cells.Cytogenet Genome Res. 2004;104(1-4):116-22. doi: 10.1159/000077475. Cytogenet Genome Res. 2004. PMID: 15162024 Review.
-
DNA damage pathways and B-cell lymphomagenesis.Curr Opin Hematol. 2018 Jul;25(4):315-322. doi: 10.1097/MOH.0000000000000433. Curr Opin Hematol. 2018. PMID: 29702521 Review.
Cited by
-
A novel homozygous C-terminal deletion in BTG4 causes zygotic cleavage failure and female infertility.J Assist Reprod Genet. 2023 Jan;40(1):75-81. doi: 10.1007/s10815-022-02664-0. Epub 2022 Dec 6. J Assist Reprod Genet. 2023. PMID: 36471203 Free PMC article.
-
Maternal effect genes as risk factors for congenital heart defects.HGG Adv. 2022 Mar 9;3(2):100098. doi: 10.1016/j.xhgg.2022.100098. eCollection 2022 Apr 14. HGG Adv. 2022. PMID: 35345810 Free PMC article.
-
Hyperthyroidism Associated with Gestational Trophoblastic Neoplasia: Systematic Literature Review and Pathways Analysis.Cancers (Basel). 2025 Apr 22;17(9):1398. doi: 10.3390/cancers17091398. Cancers (Basel). 2025. PMID: 40361325 Free PMC article. Review.
-
Revealing the expression profile of genes that encode the Subcortical Maternal Complex in human reproductive failures.Genet Mol Biol. 2023 Dec 11;46(3 Suppl 1):e20230141. doi: 10.1590/1678-4685-GMB-2023-0141. eCollection 2023. Genet Mol Biol. 2023. PMID: 38091268 Free PMC article.
-
Biallelic mutations in MOS cause female infertility characterized by human early embryonic arrest and fragmentation.EMBO Mol Med. 2021 Dec 7;13(12):e14887. doi: 10.15252/emmm.202114887. Epub 2021 Nov 15. EMBO Mol Med. 2021. PMID: 34779126 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
