

Complement Inhibitors in Clinical Trials for Glomerular Diseases
- PMID: 31611870
- PMCID: PMC6776600
- DOI: 10.3389/fimmu.2019.02166
Complement Inhibitors in Clinical Trials for Glomerular Diseases
Abstract
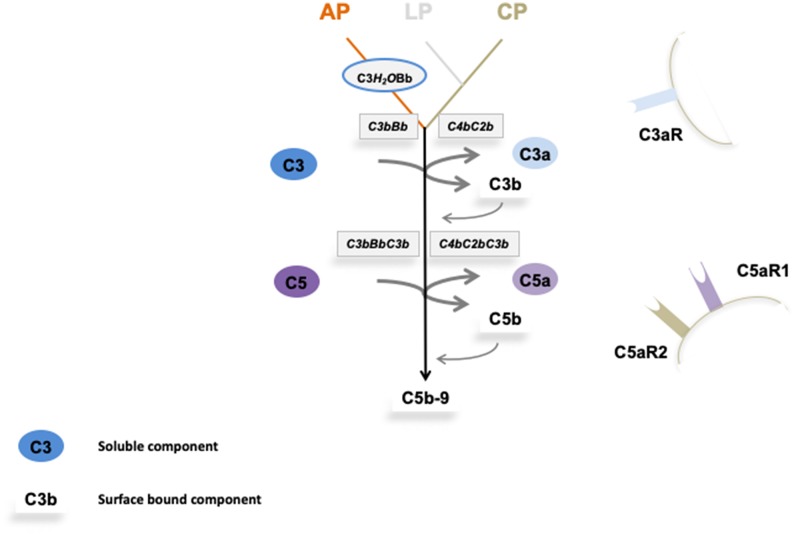
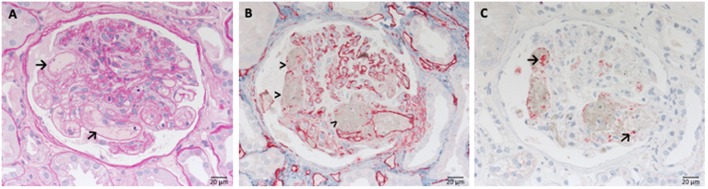
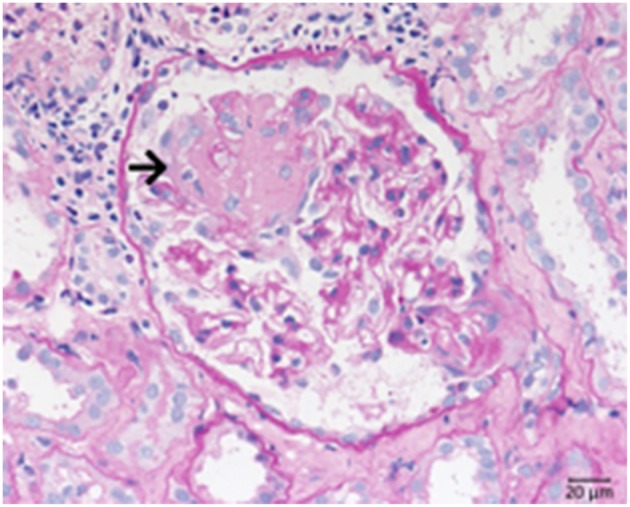
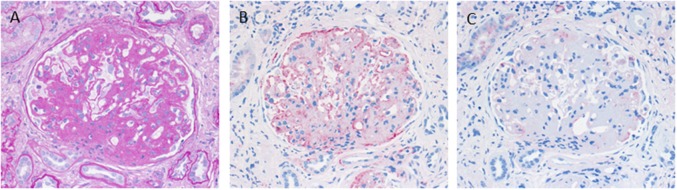
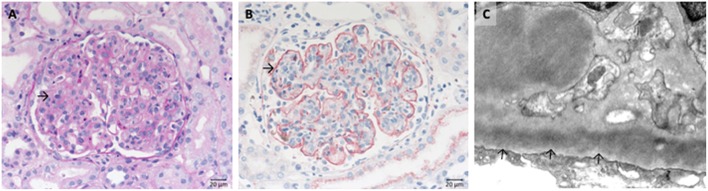
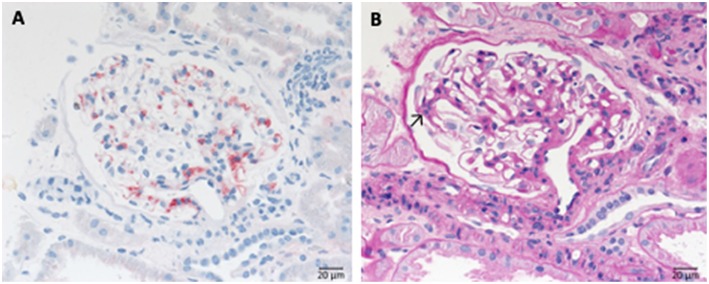
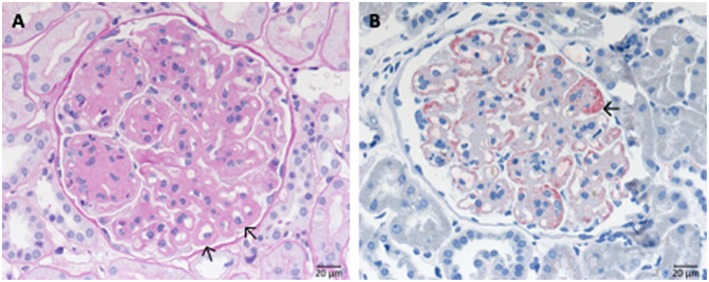
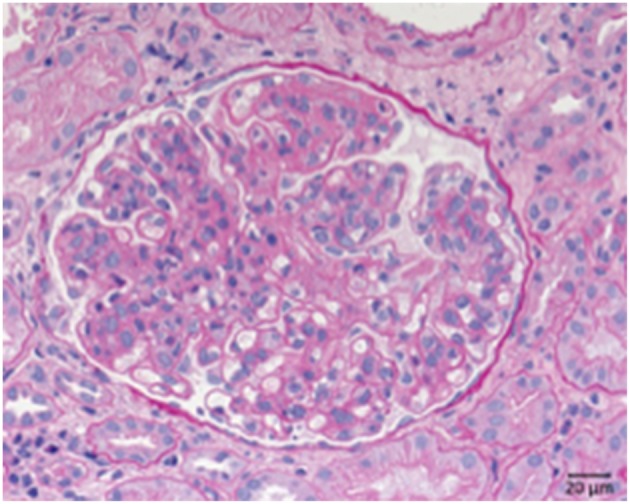
Defective complement action is a cause of several human glomerular diseases including atypical hemolytic uremic syndrome (aHUS), anti-neutrophil cytoplasmic antibody mediated vasculitis (ANCA), C3 glomerulopathy, IgA nephropathy, immune complex membranoproliferative glomerulonephritis, ischemic reperfusion injury, lupus nephritis, membranous nephropathy, and chronic transplant mediated glomerulopathy. Here we summarize ongoing clinical trials of complement inhibitors in nine glomerular diseases and show which inhibitors are used in trials for these renal disorders (http://clinicaltrials.gov).
Keywords: ANCA; C3 glomerulopathy; aHUS; clinical trials; complement; glomerular disease; inhibitors.
Copyright © 2019 Zipfel, Wiech, Rudnick, Afonso, Person and Skerka.
Figures
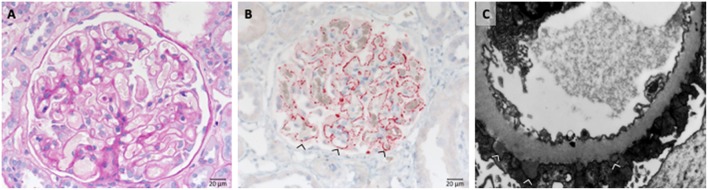
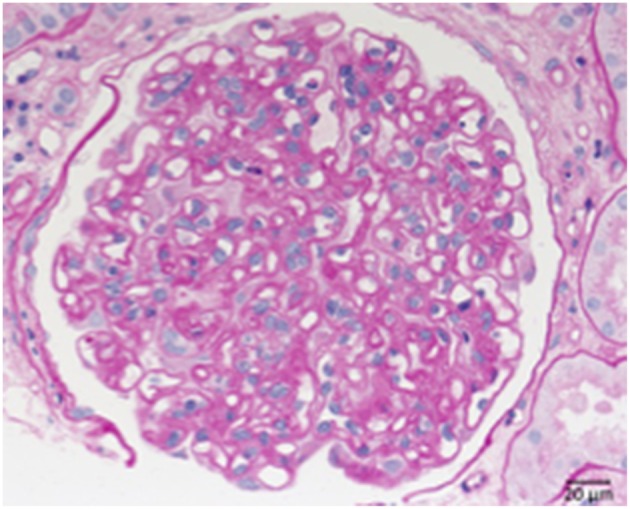
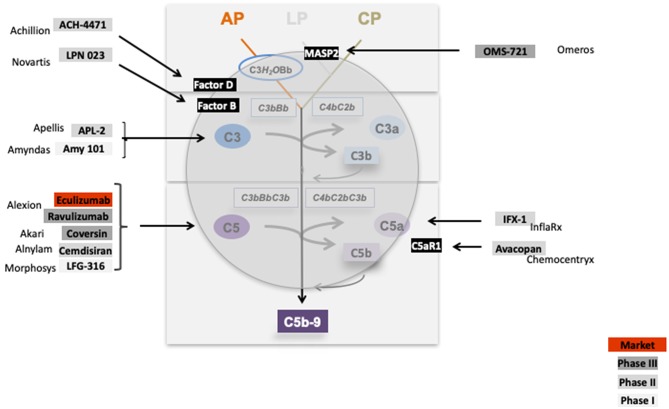
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous