

Panax Notoginseng Saponins Protect Cardiac Myocytes Against Endoplasmic Reticulum Stress and Associated Apoptosis Through Mediation of Intracellular Calcium Homeostasis
- PMID: 31616293
- PMCID: PMC6764115
- DOI: 10.3389/fphar.2019.01013
Panax Notoginseng Saponins Protect Cardiac Myocytes Against Endoplasmic Reticulum Stress and Associated Apoptosis Through Mediation of Intracellular Calcium Homeostasis
Abstract
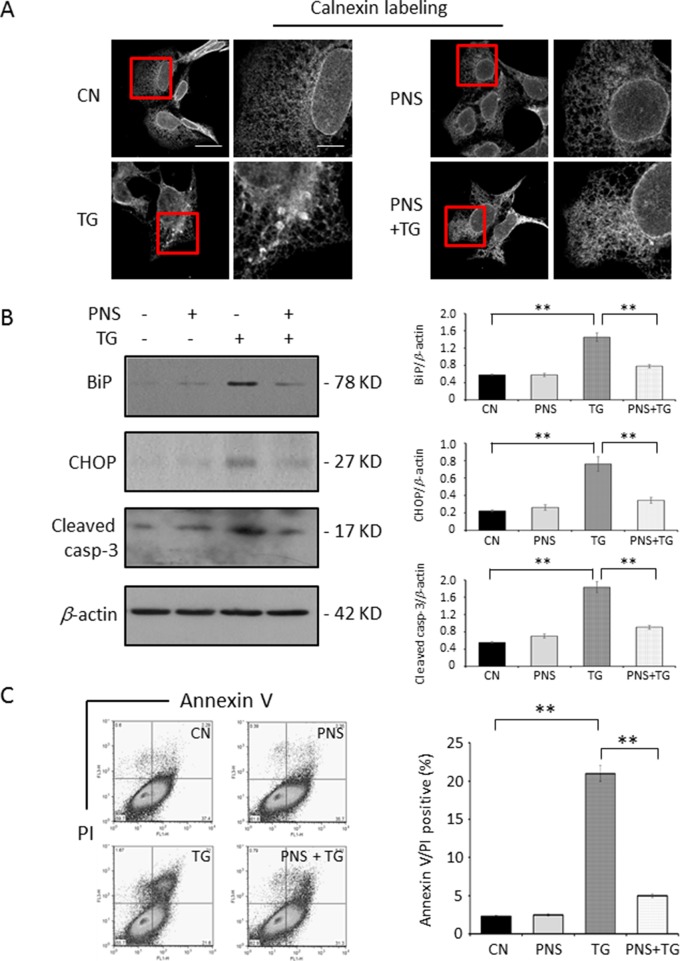
Endoplasmic reticulum (ER) stress has been demonstrated to play important roles in the pathogenesis of various cardiovascular diseases. The ER stress pathway is therefore a promising therapeutic target in cardiovascular disease. Although Panax notoginseng saponins (PNS) are one of the patent medicines that are traditionally used to treat cardiovascular disorders, their effects on ER stress in cardiac myocytes remain unexploited so far. This study investigates the effects of PNS on ER stress and its associated cell apoptosis along with the related mechanism in cardiac myocytes. PNS compounds were identified via high-performance liquid chromatograph (HPLC) assay. PNS-pretreated H9c2 cells, HL-1 cells, and primary cultured neonatal rat cardiomyocytes were stimulated with thapsigargin (TG) to induce ER stress response and apoptosis. ER stress response was tested by immunofluorescence or immunoblot of the ER protein chaperones-calnexin, binding immunoglobulin protein (BiP) and the C/EBP homologous protein (CHOP). Cell viability was tested by methyl thiazolyl tetrazolium (MTT) assay. Cell apoptosis was detected by immunoblot of Cleaved caspase-3 and flow cytometry analysis of Annexin V/propidium iodide (PI) staining. Cytosolic, mitochondrial, and ER calcium dynamics were investigated by calcium imaging. Moreover, a ryanodine receptor type-2 (RyR2) overexpression stable cell line was generated to verify the mechanism of RyR2 involved in PNS in the inhibition of ER stress and cell apoptosis. We demonstrate here that PNS protected cardiac myocytes from ER stress response and associated cell death in a concentration-dependent manner. Importantly, PNS reduced the elevation of cytosolic calcium, mitochondria calcium, as well as ER calcium in response to either TG or histamine treatment. PNS protection in ER stress was regulated by RyR2 expression. In summary, PNS protection against TG-induced ER stress response and its associated cell apoptosis in cardiac myocytes is calcium dependent. Through the regulation of ER calcium release mediated by RyR2, a novel mechanism for PNS in the prevention of cardiovascular diseases is thereby identified.
Keywords: Panax notoginseng saponins; apoptosis; endoplasmic reticulum stress; intracellular calcium homeostasis; ryanodine receptor.
Copyright © 2019 Chen, Xue, Li, Xiao, Shangguan, Zhang, Bai, Liu and Li.
Figures







References
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous