

The ADP/ATP translocase drives mitophagy independent of nucleotide exchange
- PMID: 31618756
- PMCID: PMC6858570
- DOI: 10.1038/s41586-019-1667-4
The ADP/ATP translocase drives mitophagy independent of nucleotide exchange
Abstract
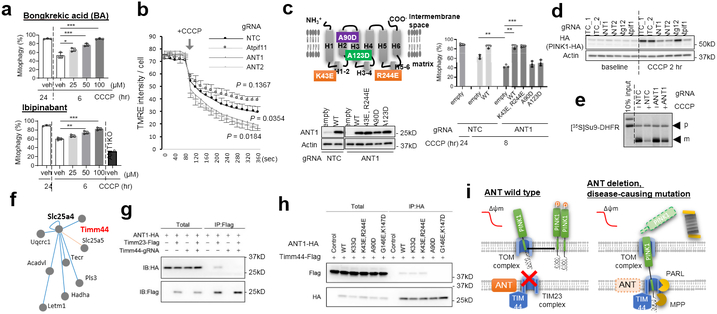
Mitochondrial homeostasis depends on mitophagy, the programmed degradation of mitochondria. Only a few proteins are known to participate in mitophagy. Here we develop a multidimensional CRISPR-Cas9 genetic screen, using multiple mitophagy reporter systems and pro-mitophagy triggers, and identify numerous components of parkin-dependent mitophagy1. Unexpectedly, we find that the adenine nucleotide translocator (ANT) complex is required for mitophagy in several cell types. Whereas pharmacological inhibition of ANT-mediated ADP/ATP exchange promotes mitophagy, genetic ablation of ANT paradoxically suppresses mitophagy. Notably, ANT promotes mitophagy independently of its nucleotide translocase catalytic activity. Instead, the ANT complex is required for inhibition of the presequence translocase TIM23, which leads to stabilization of PINK1, in response to bioenergetic collapse. ANT modulates TIM23 indirectly via interaction with TIM44, which regulates peptide import through TIM232. Mice that lack ANT1 show blunted mitophagy and consequent profound accumulation of aberrant mitochondria. Disease-causing human mutations in ANT1 abrogate binding to TIM44 and TIM23 and inhibit mitophagy. Together, our findings show that ANT is an essential and fundamental mediator of mitophagy in health and disease.
Figures









References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
