

Microbe-host interplay in atopic dermatitis and psoriasis
- PMID: 31619666
- PMCID: PMC6795799
- DOI: 10.1038/s41467-019-12253-y
Microbe-host interplay in atopic dermatitis and psoriasis
Abstract
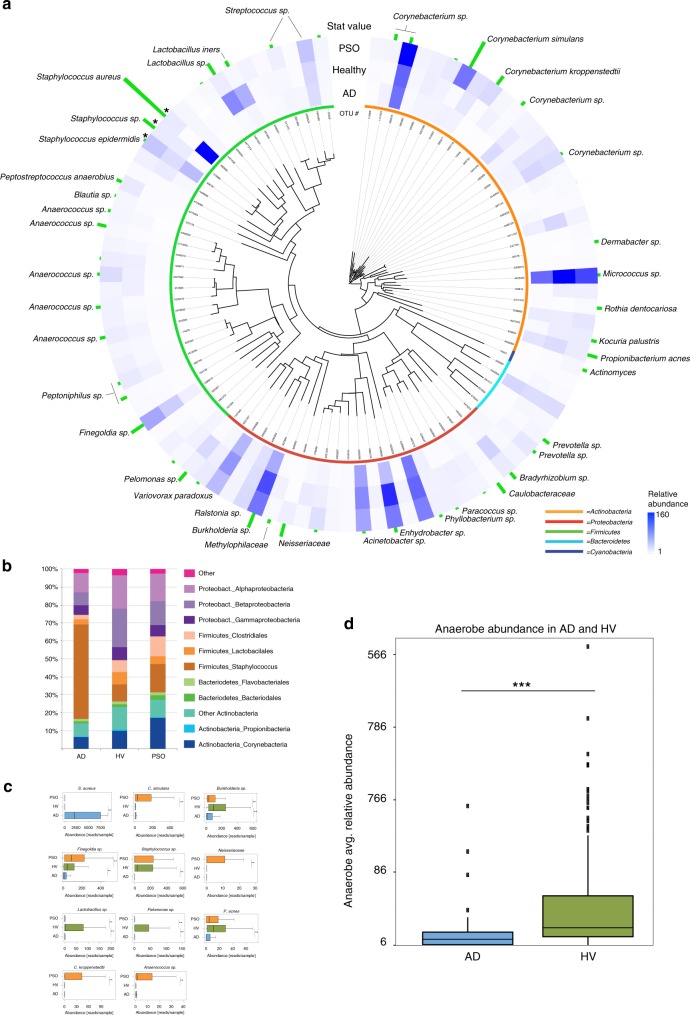
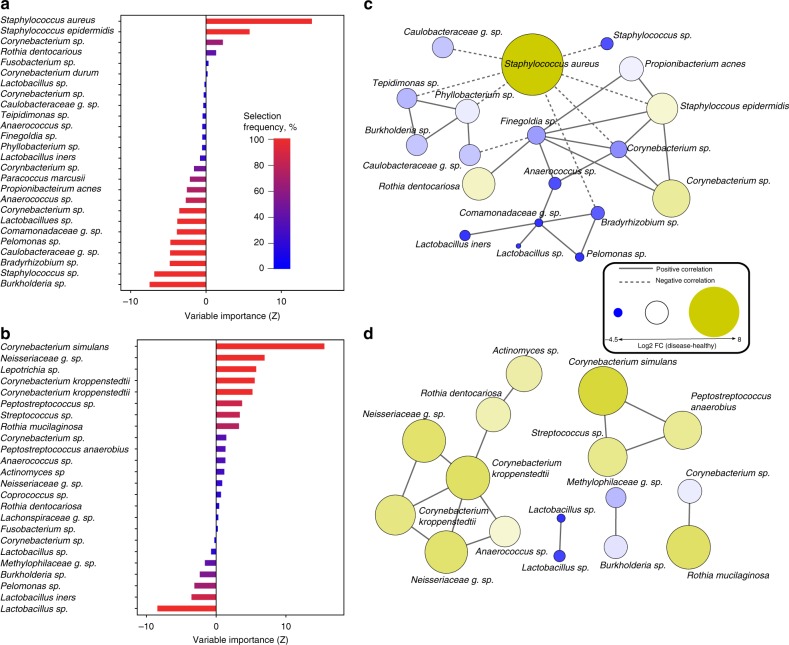
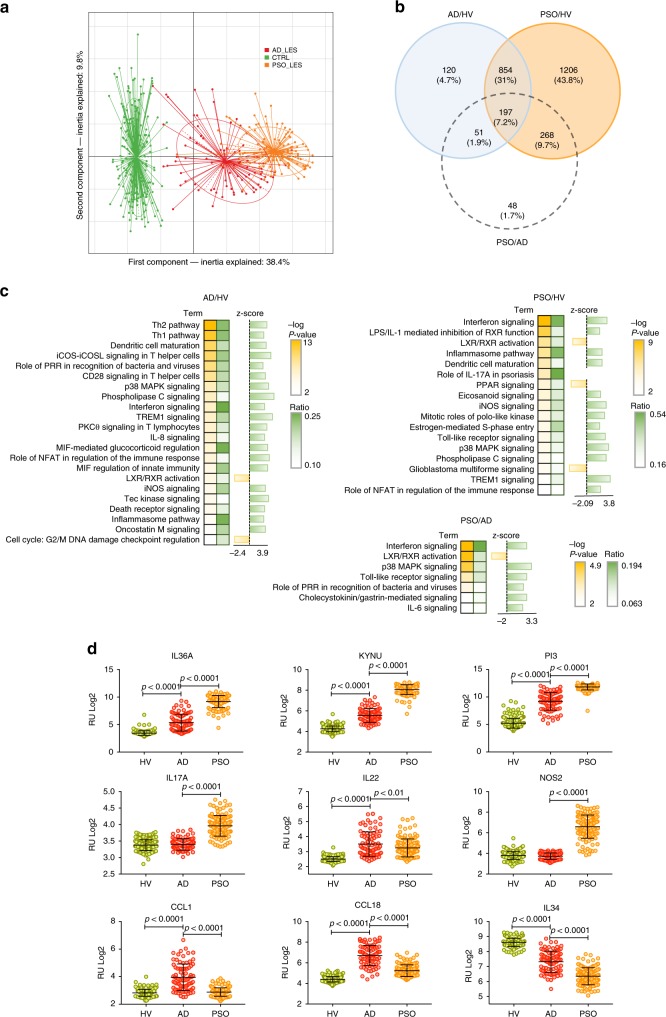
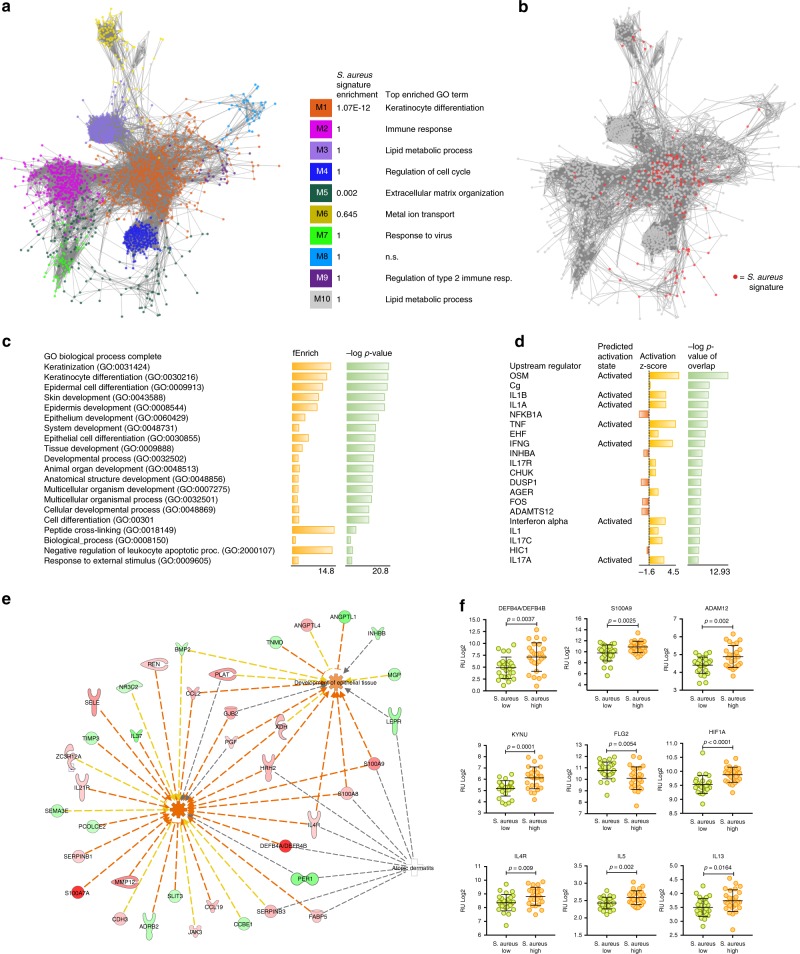
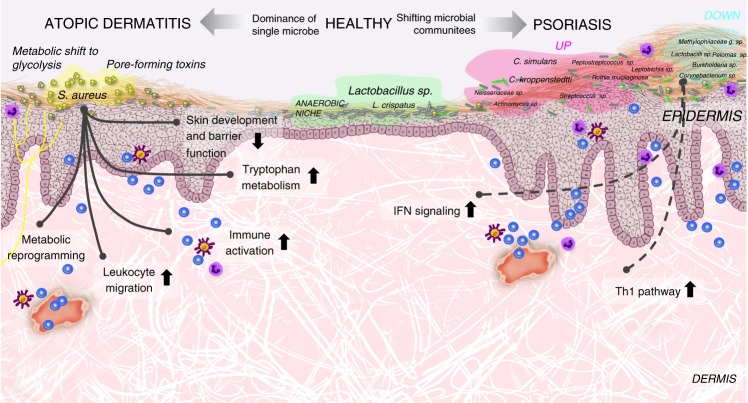
Despite recent advances in understanding microbial diversity in skin homeostasis, the relevance of microbial dysbiosis in inflammatory disease is poorly understood. Here we perform a comparative analysis of skin microbial communities coupled to global patterns of cutaneous gene expression in patients with atopic dermatitis or psoriasis. The skin microbiota is analysed by 16S amplicon or whole genome sequencing and the skin transcriptome by microarrays, followed by integration of the data layers. We find that atopic dermatitis and psoriasis can be classified by distinct microbes, which differ from healthy volunteers microbiome composition. Atopic dermatitis is dominated by a single microbe (Staphylococcus aureus), and associated with a disease relevant host transcriptomic signature enriched for skin barrier function, tryptophan metabolism and immune activation. In contrast, psoriasis is characterized by co-occurring communities of microbes with weak associations with disease related gene expression. Our work provides a basis for biomarker discovery and targeted therapies in skin dysbiosis.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
