

Prelamin A mediates myocardial inflammation in dilated and HIV-associated cardiomyopathies
- PMID: 31622279
- PMCID: PMC6948859
- DOI: 10.1172/jci.insight.126315
Prelamin A mediates myocardial inflammation in dilated and HIV-associated cardiomyopathies
Abstract
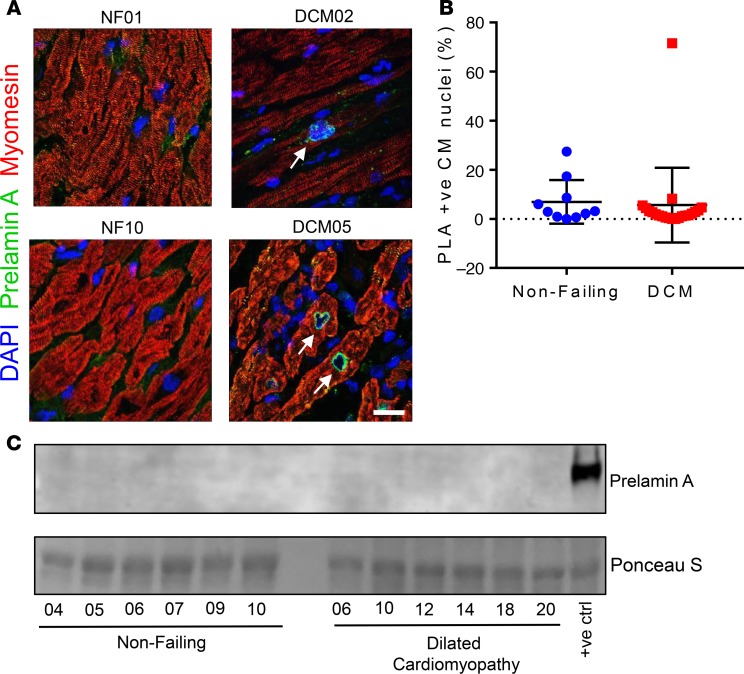
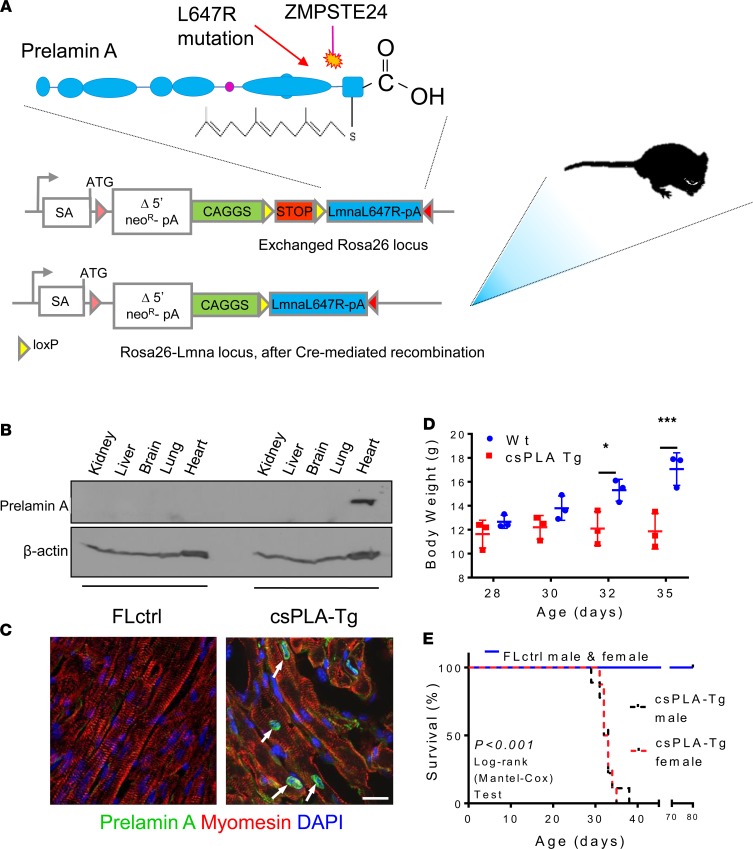
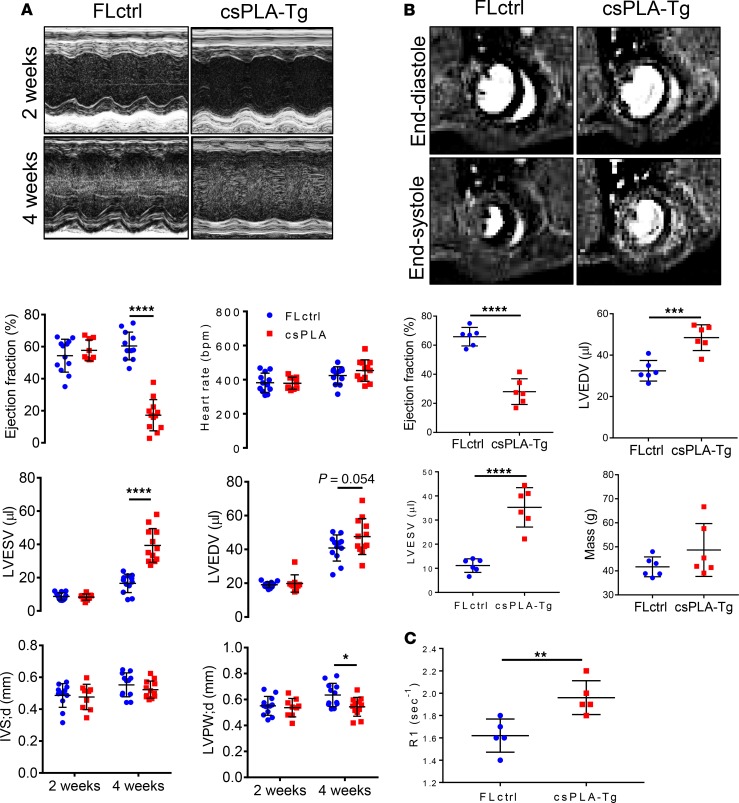
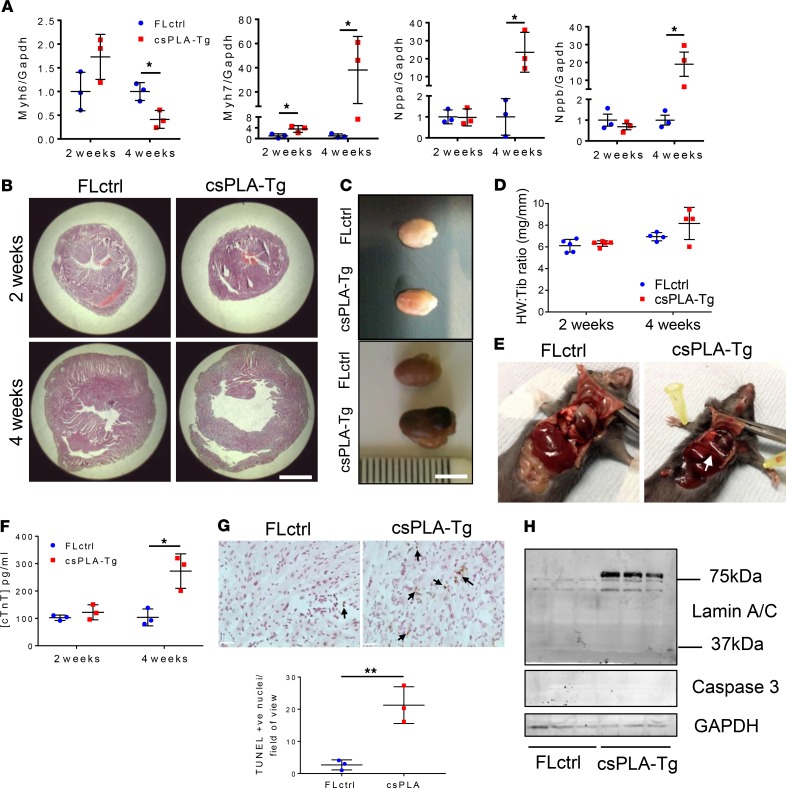
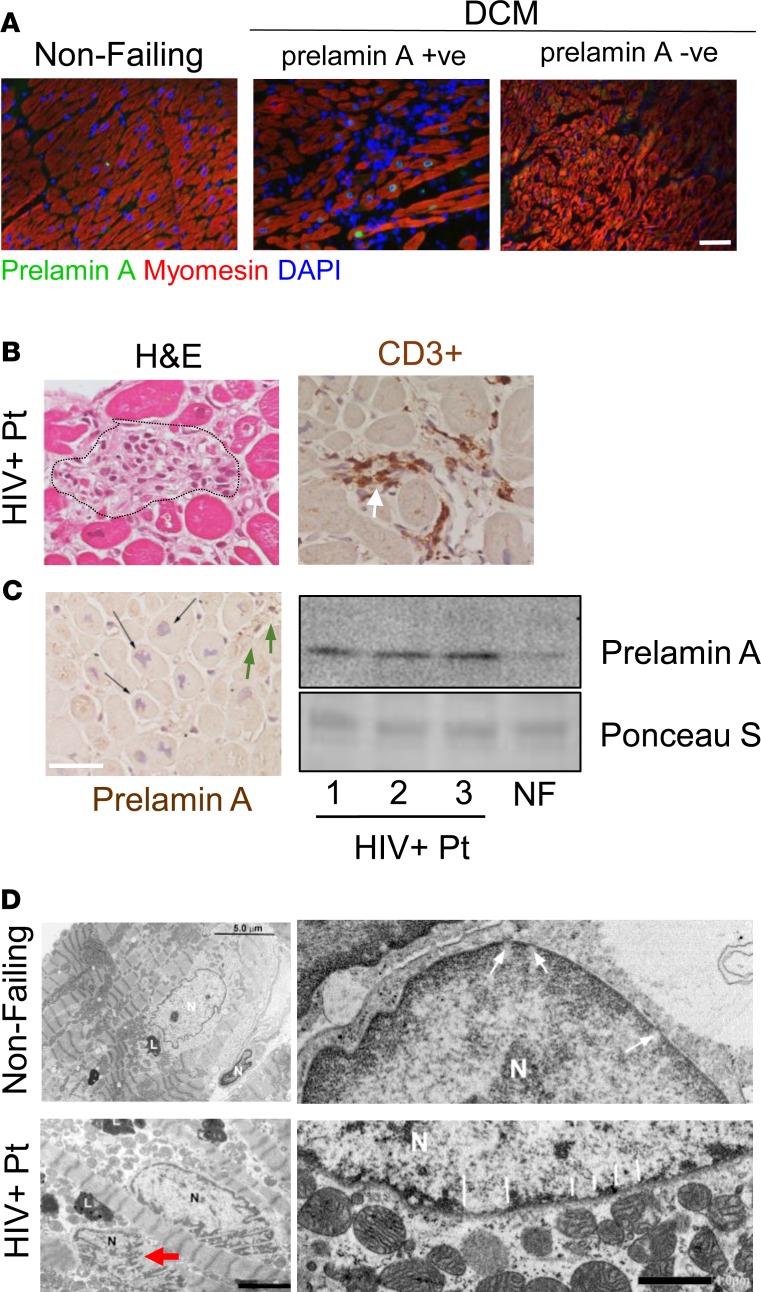
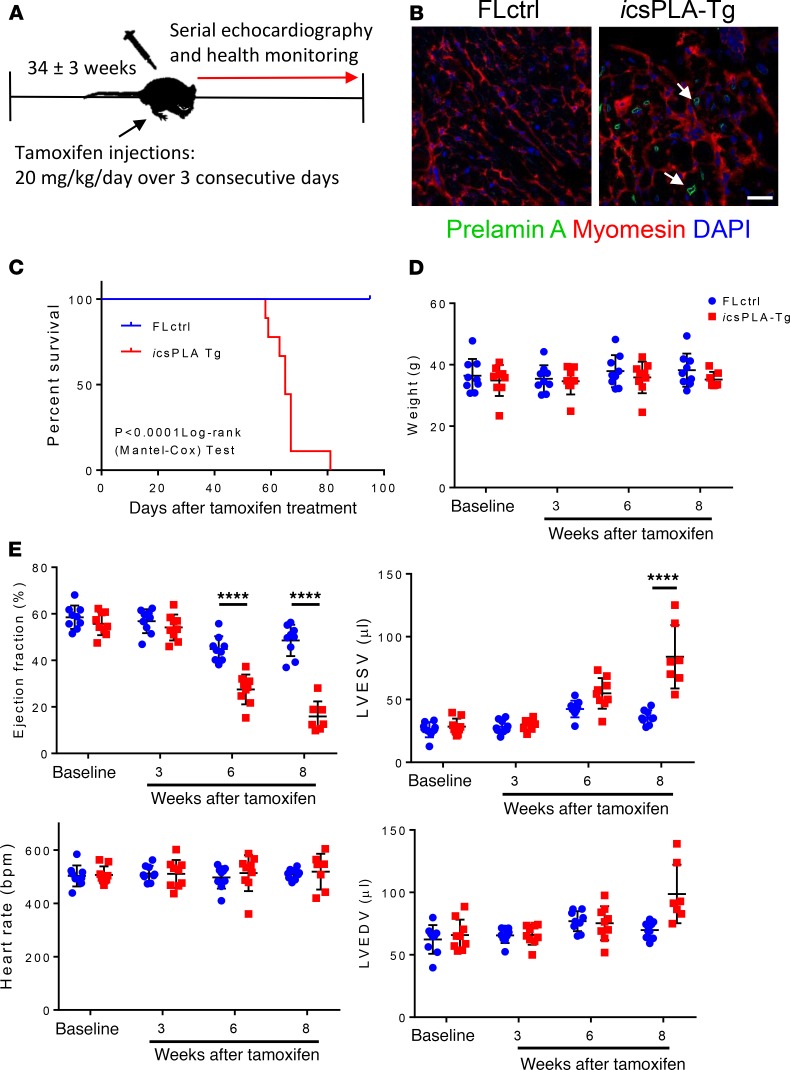
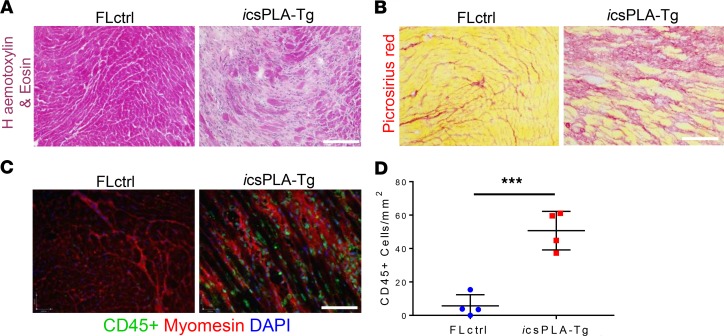
Cardiomyopathies are complex heart muscle diseases that can be inherited or acquired. Dilated cardiomyopathy can result from mutations in LMNA, encoding the nuclear intermediate filament proteins lamin A/C. Some LMNA mutations lead to accumulation of the lamin A precursor, prelamin A, which is disease causing in a number of tissues, yet its impact upon the heart is unknown. Here, we discovered myocardial prelamin A accumulation occurred in a case of dilated cardiomyopathy, and we show that a potentially novel mouse model of cardiac-specific prelamin A accumulation exhibited a phenotype consistent with inflammatory cardiomyopathy, which we observed to be similar to HIV-associated cardiomyopathy, an acquired disease state. Numerous HIV protease therapies are known to inhibit ZMPSTE24, the enzyme responsible for prelamin A processing, and we confirmed that accumulation of prelamin A occurred in HIV+ patient cardiac biopsies. These findings (a) confirm a unifying pathological role for prelamin A common to genetic and acquired cardiomyopathies; (b) have implications for the management of HIV patients with cardiac disease, suggesting protease inhibitors should be replaced with alternative therapies (i.e., nonnucleoside reverse transcriptase inhibitors); and (c) suggest that targeting inflammation may be a useful treatment strategy for certain forms of inherited cardiomyopathy.
Keywords: AIDS/HIV; Cardiology; Cardiovascular disease.
Conflict of interest statement
Figures
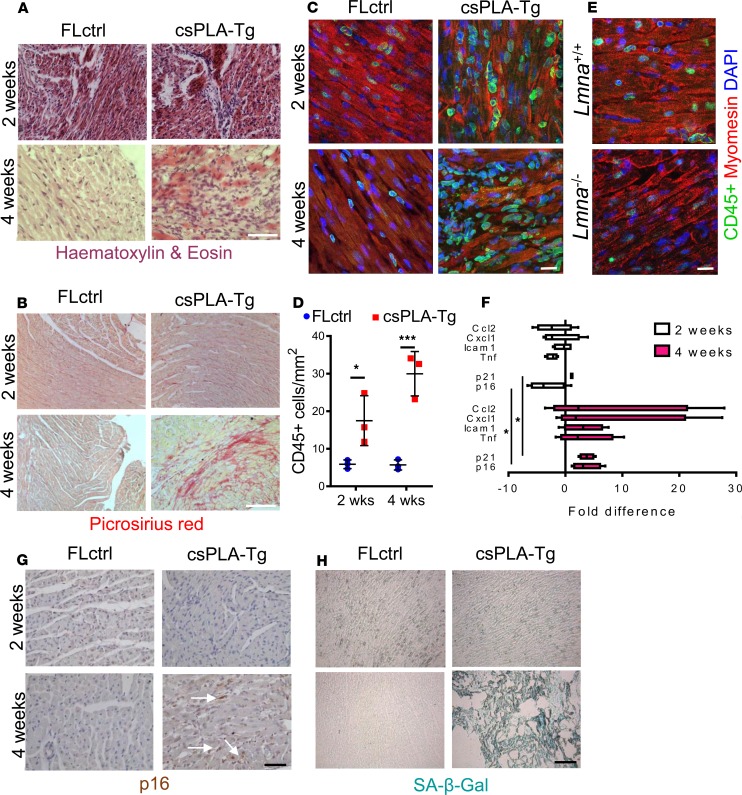
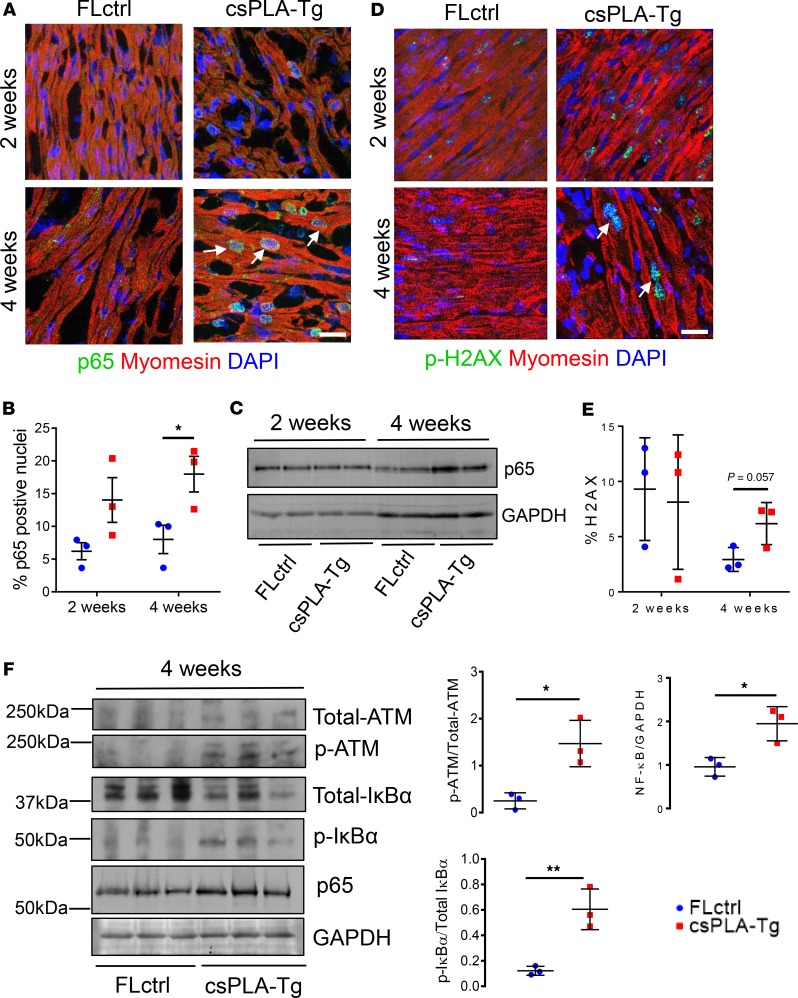
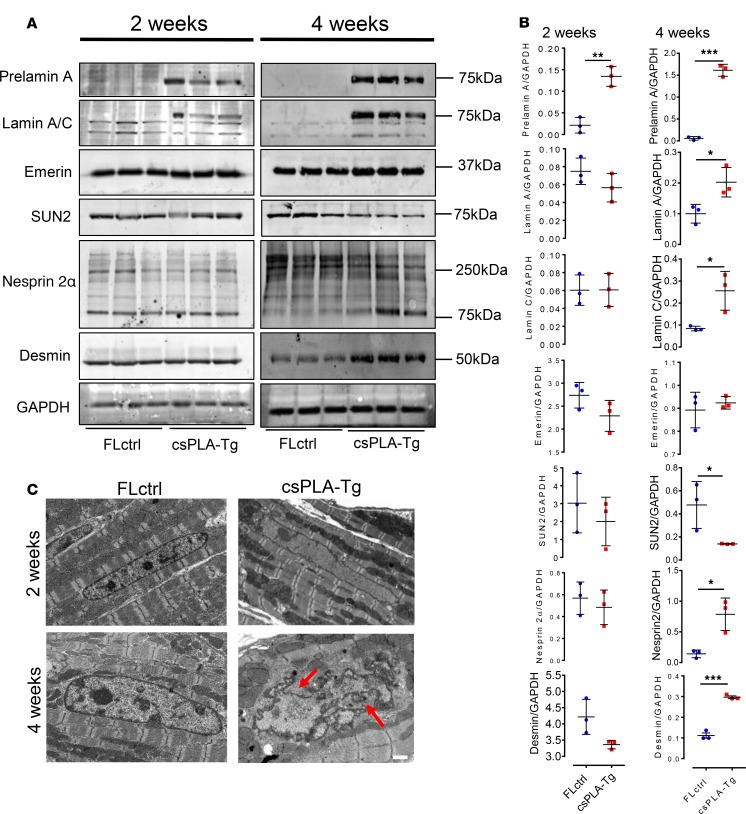
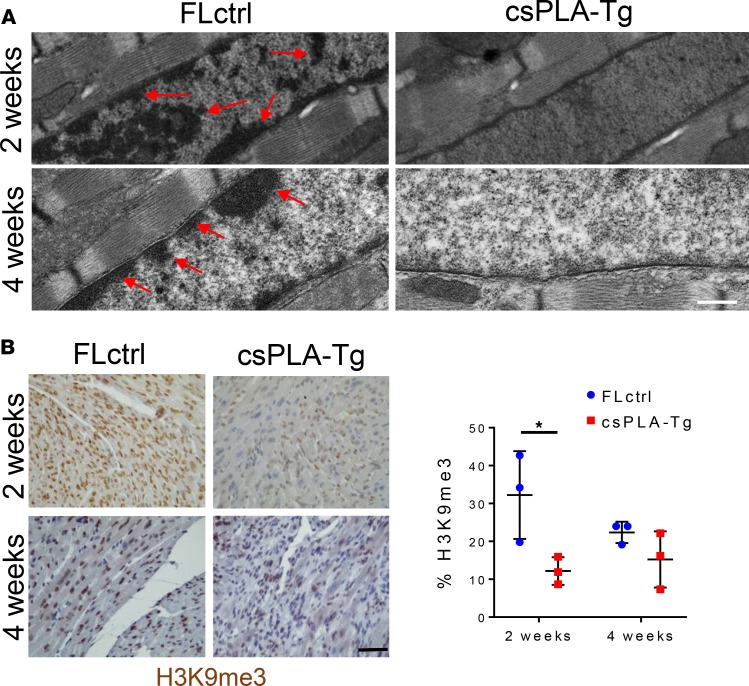
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
