

Single-cell transcriptomics of human T cells reveals tissue and activation signatures in health and disease
- PMID: 31624246
- PMCID: PMC6797728
- DOI: 10.1038/s41467-019-12464-3
Single-cell transcriptomics of human T cells reveals tissue and activation signatures in health and disease
Abstract
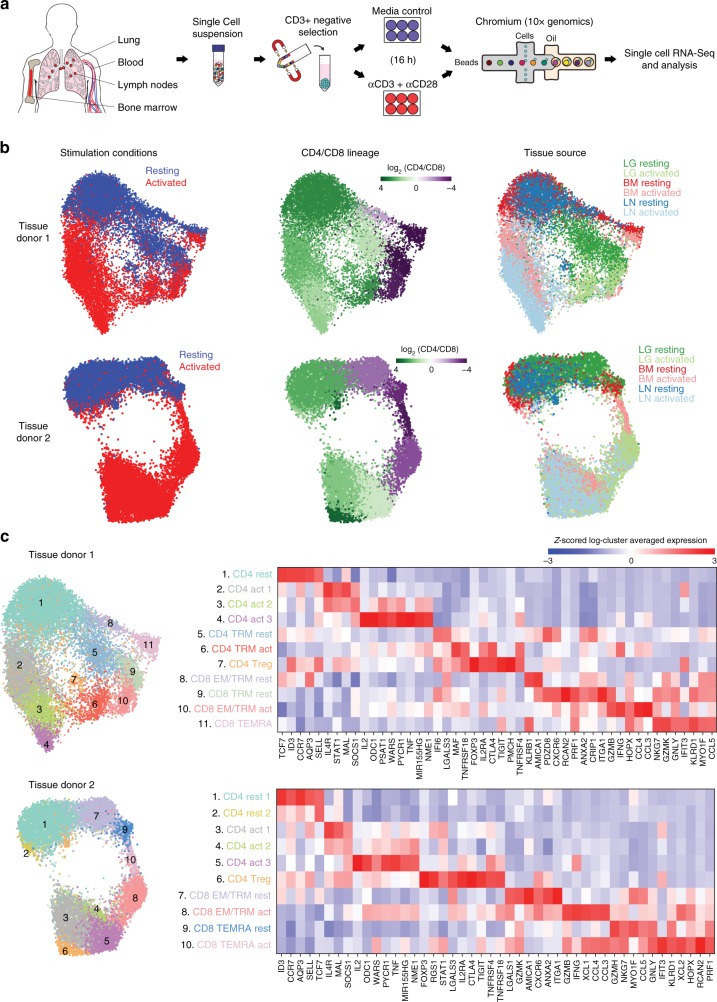
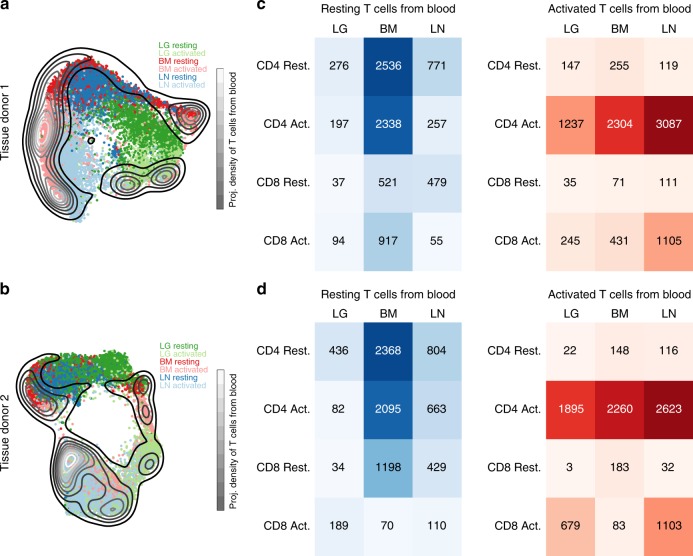
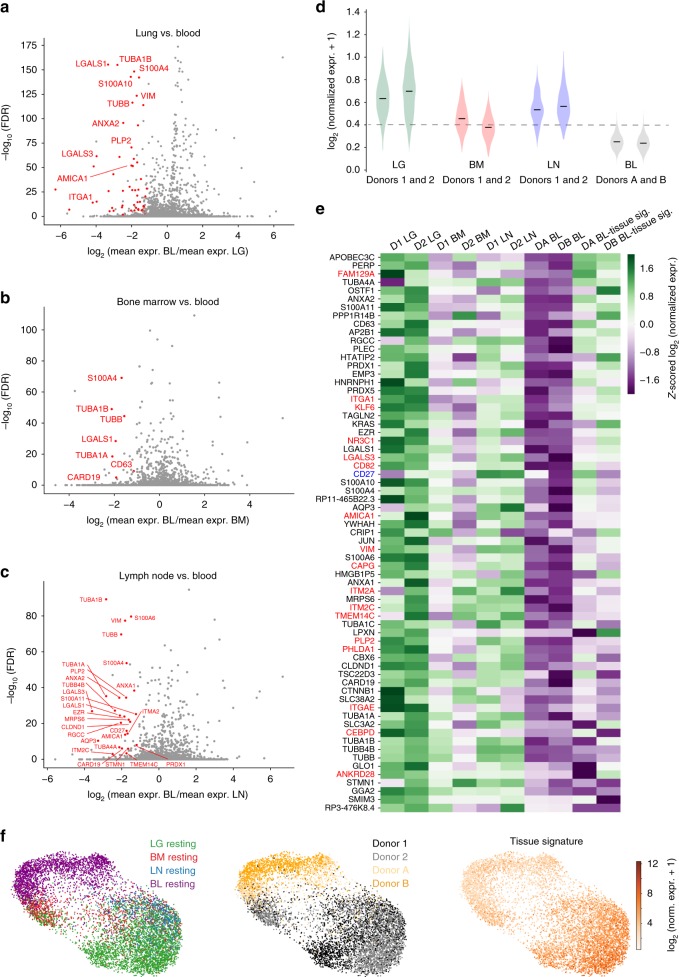
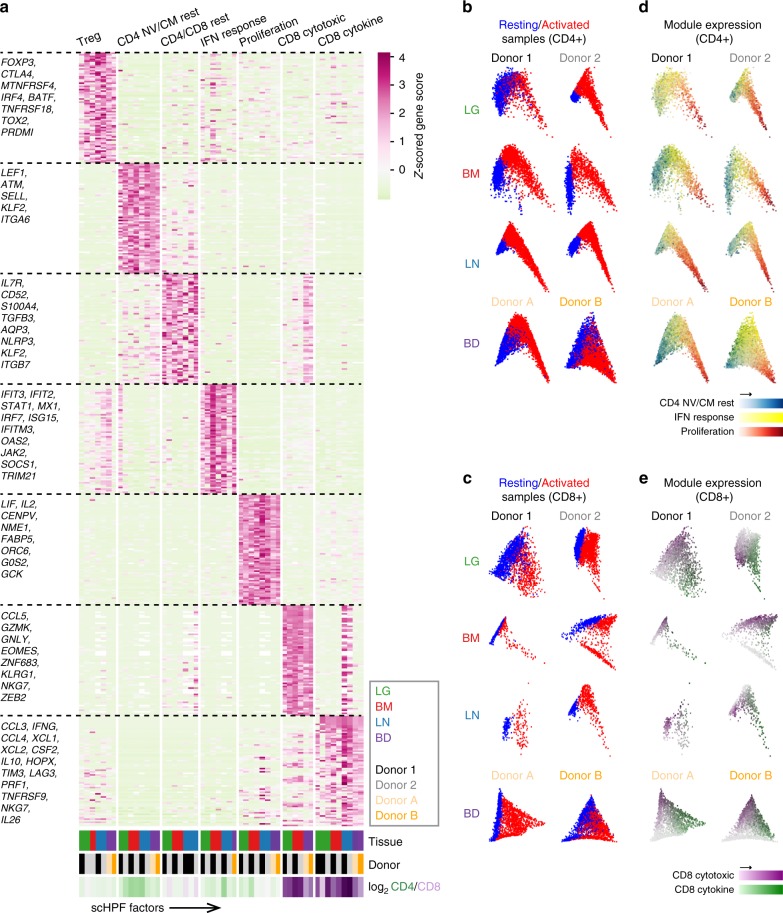
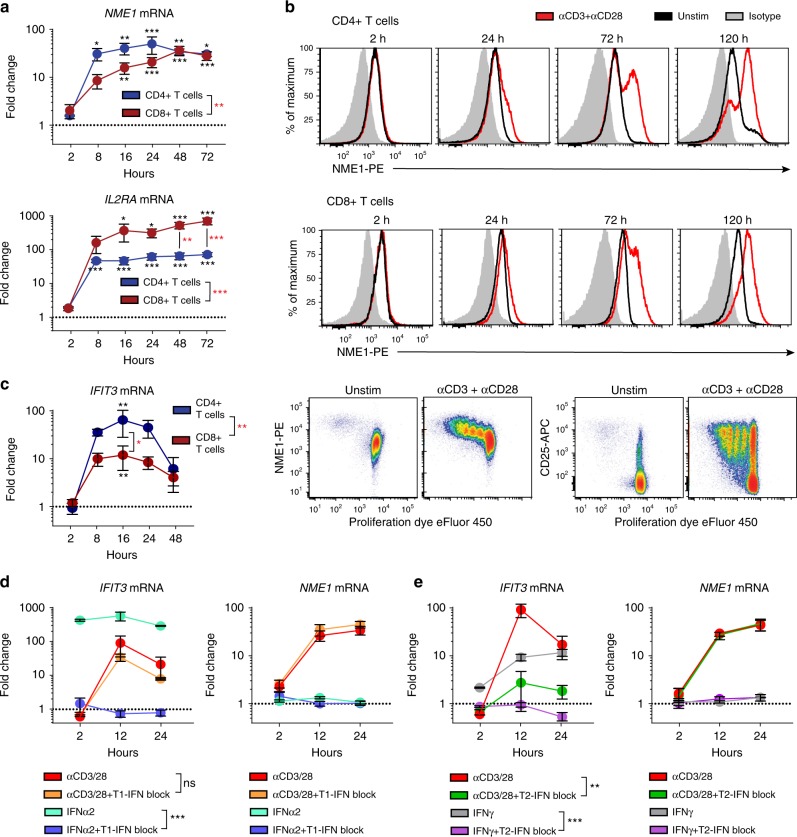
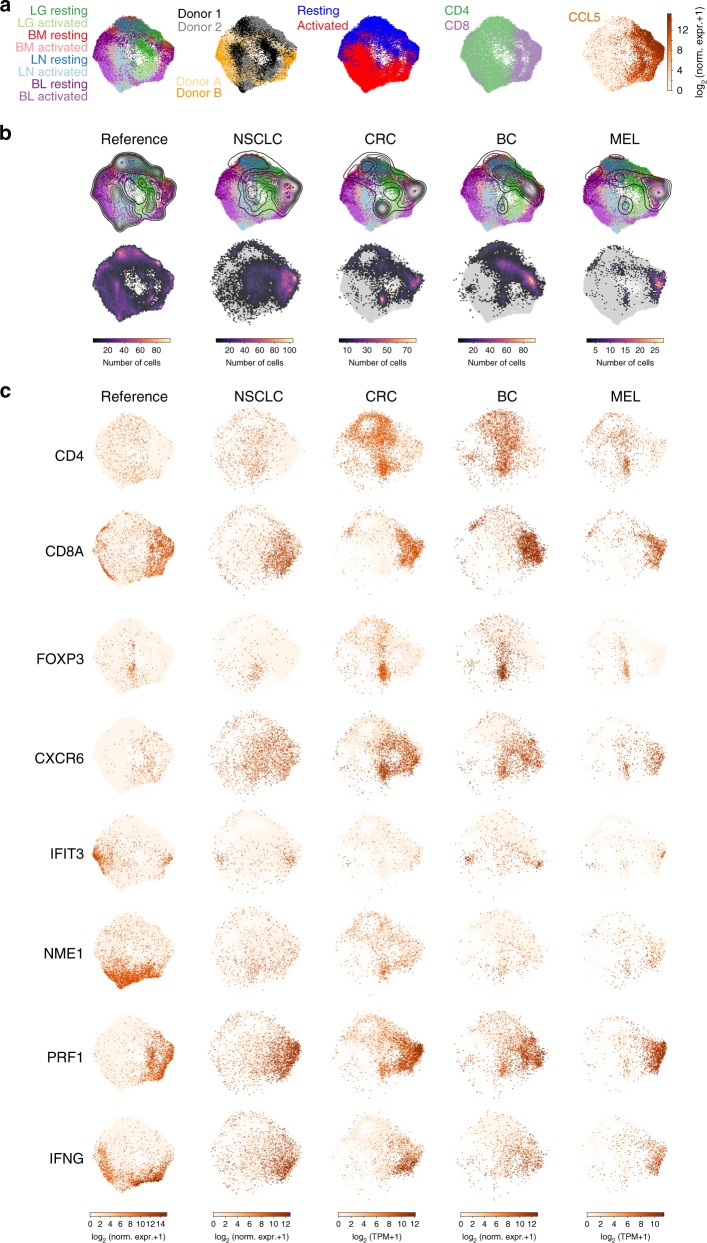
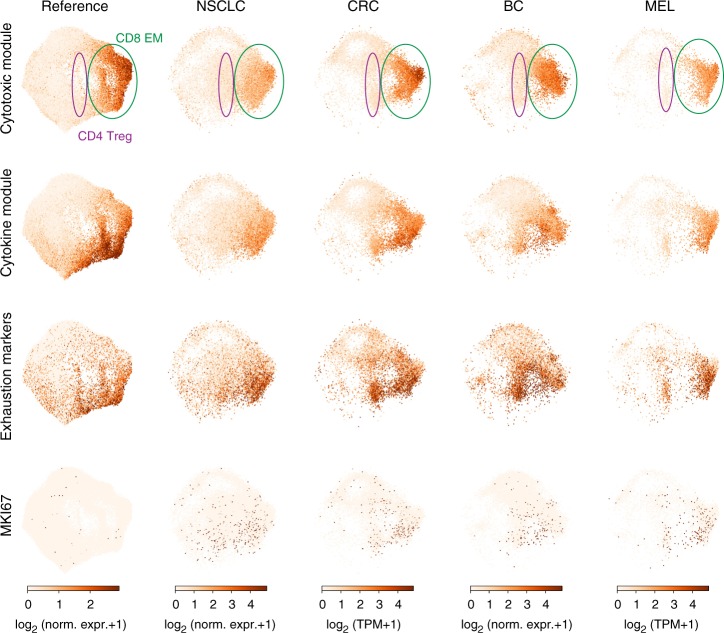
Human T cells coordinate adaptive immunity in diverse anatomic compartments through production of cytokines and effector molecules, but it is unclear how tissue site influences T cell persistence and function. Here, we use single cell RNA-sequencing (scRNA-seq) to define the heterogeneity of human T cells isolated from lungs, lymph nodes, bone marrow and blood, and their functional responses following stimulation. Through analysis of >50,000 resting and activated T cells, we reveal tissue T cell signatures in mucosal and lymphoid sites, and lineage-specific activation states across all sites including distinct effector states for CD8+ T cells and an interferon-response state for CD4+ T cells. Comparing scRNA-seq profiles of tumor-associated T cells to our dataset reveals predominant activated CD8+ compared to CD4+ T cell states within multiple tumor types. Our results therefore establish a high dimensional reference map of human T cell activation in health for analyzing T cells in disease.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
