

De novo and recessive forms of congenital heart disease have distinct genetic and phenotypic landscapes
- PMID: 31624253
- PMCID: PMC6797711
- DOI: 10.1038/s41467-019-12582-y
De novo and recessive forms of congenital heart disease have distinct genetic and phenotypic landscapes
Abstract
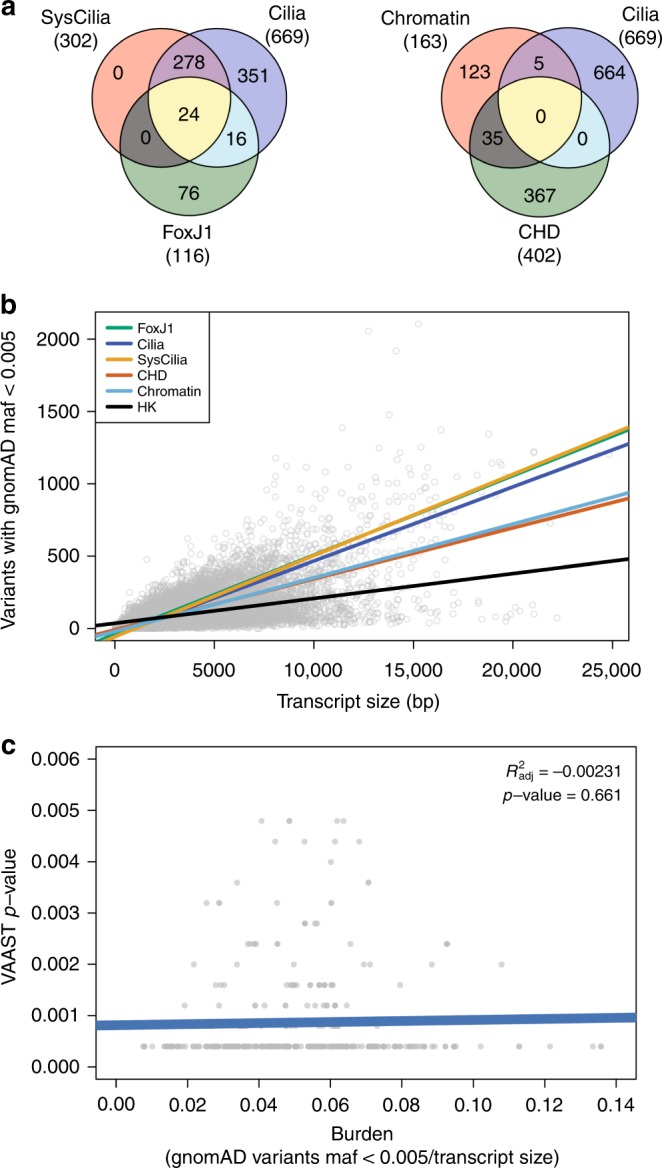
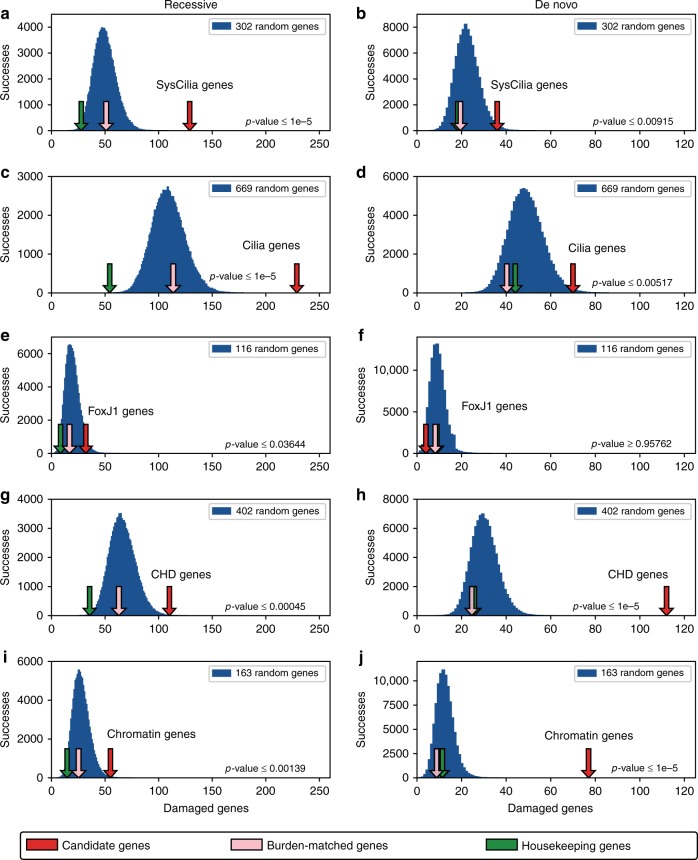
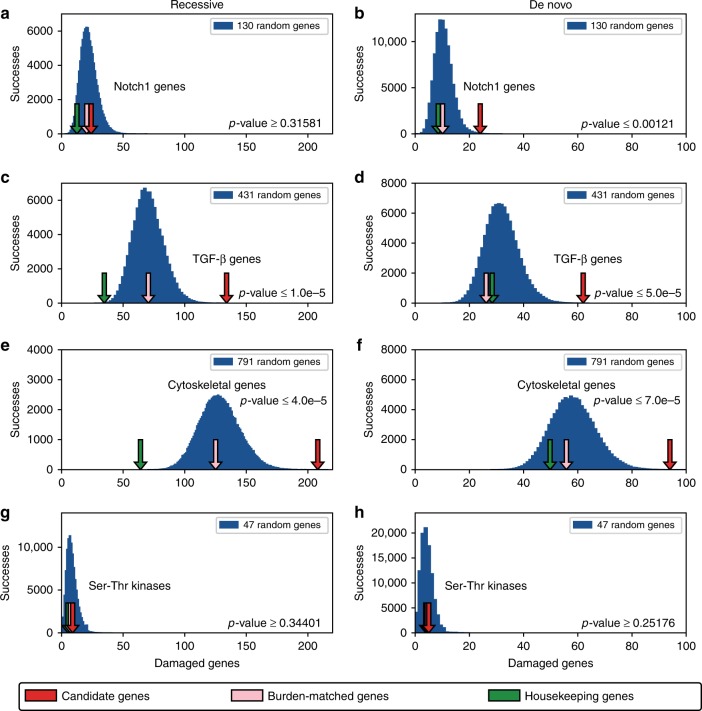
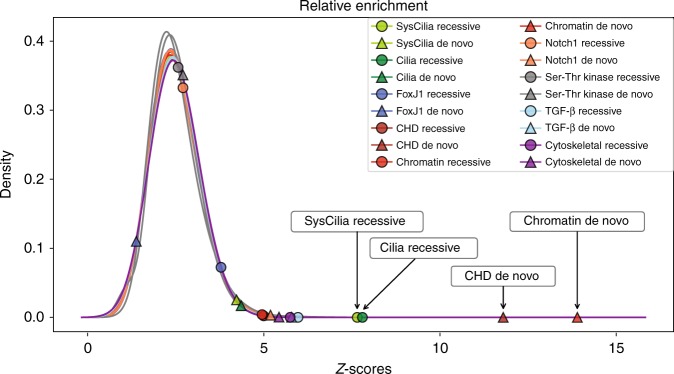
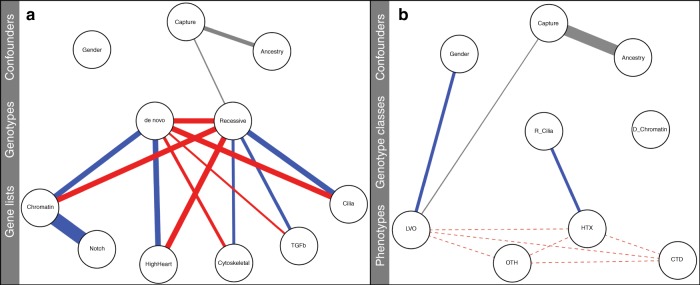
The genetic architecture of sporadic congenital heart disease (CHD) is characterized by enrichment in damaging de novo variants in chromatin-modifying genes. To test the hypothesis that gene pathways contributing to de novo forms of CHD are distinct from those for recessive forms, we analyze 2391 whole-exome trios from the Pediatric Cardiac Genomics Consortium. We deploy a permutation-based gene-burden analysis to identify damaging recessive and compound heterozygous genotypes and disease genes, controlling for confounding effects, such as background mutation rate and ancestry. Cilia-related genes are significantly enriched for damaging rare recessive genotypes, but comparatively depleted for de novo variants. The opposite trend is observed for chromatin-modifying genes. Other cardiac developmental gene classes have less stratification by mode of inheritance than cilia and chromatin-modifying gene classes. Our analyses reveal dominant and recessive CHD are associated with distinct gene functions, with cilia-related genes providing a reservoir of rare segregating variation leading to CHD.
Conflict of interest statement
The authors declare the following competing interests: VAAST and PHEVOR have been commercialized by Fabric Genomics, Inc. (to M.Y.).
Figures

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
