

Tools for fundamental analysis functions of TCR repertoires: a systematic comparison
- PMID: 31624828
- PMCID: PMC7947996
- DOI: 10.1093/bib/bbz092
Tools for fundamental analysis functions of TCR repertoires: a systematic comparison
Abstract
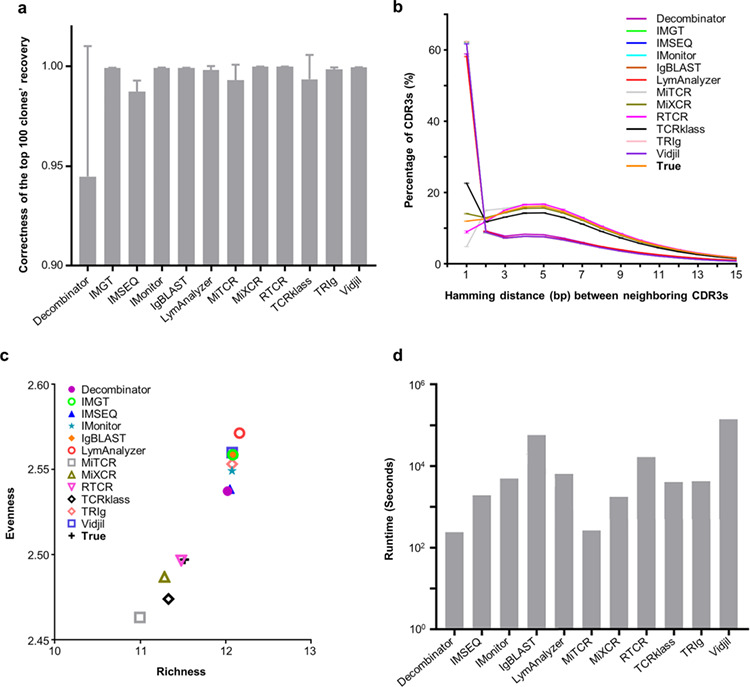
The full set of T cell receptors (TCRs) in an individual is known as his or her TCR repertoire. Defining TCR repertoires under physiological conditions and in response to a disease or vaccine may lead to a better understanding of adaptive immunity and thus has great biological and clinical value. In the past decade, several high-throughput sequencing-based tools have been developed to assign TCRs to germline genes and to extract complementarity-determining region 3 (CDR3) sequences using different algorithms. Although these tools claim to be able to perform the full range of fundamental TCR repertoire analyses, there is no clear consensus of which tool is best suited to particular projects. Here, we present a systematic analysis of 12 available TCR repertoire analysis tools using simulated data, with an emphasis on fundamental analysis functions. Our results shed light on the detailed functions of TCR repertoire analysis tools and may therefore help researchers in the field to choose the right tools for their particular experimental design.
Keywords: in silico simulation; T-cell receptor repertoire; high-throughput sequencing; immunology; tools benchmarking.
© The Author(s) 2019. Published by Oxford University Press.
Figures
References
-
- Nikolich-Žugich J, Slifka MK, Messaoudi I. The many important facets of T-cell repertoire diversity. Nat Rev Immuno 2004;4:123–32. - PubMed
-
- Dahal-Koirala S, Risnes LF, Christophersen A, et al. TCR sequencing of single cells reactive to DQ2.5-glia-α2 and DQ2.5-glia-ω2 reveals clonal expansion and epitope-specific V-gene usage. Mucosal Immunol 2016;9:587–96. - PubMed
-
- Delemarre EM, van den Broek T, Mijnheer G, et al. Autologous stem cell transplantation aids autoimmune patients by functional renewal and TCR diversification of regulatory T cells. Blood 2016;127:91–101. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
