

MicroRNA-134-5p inhibition rescues long-term plasticity and synaptic tagging/capture in an Aβ(1-42)-induced model of Alzheimer's disease
- PMID: 31625272
- PMCID: PMC6974725
- DOI: 10.1111/acel.13046
MicroRNA-134-5p inhibition rescues long-term plasticity and synaptic tagging/capture in an Aβ(1-42)-induced model of Alzheimer's disease
Abstract
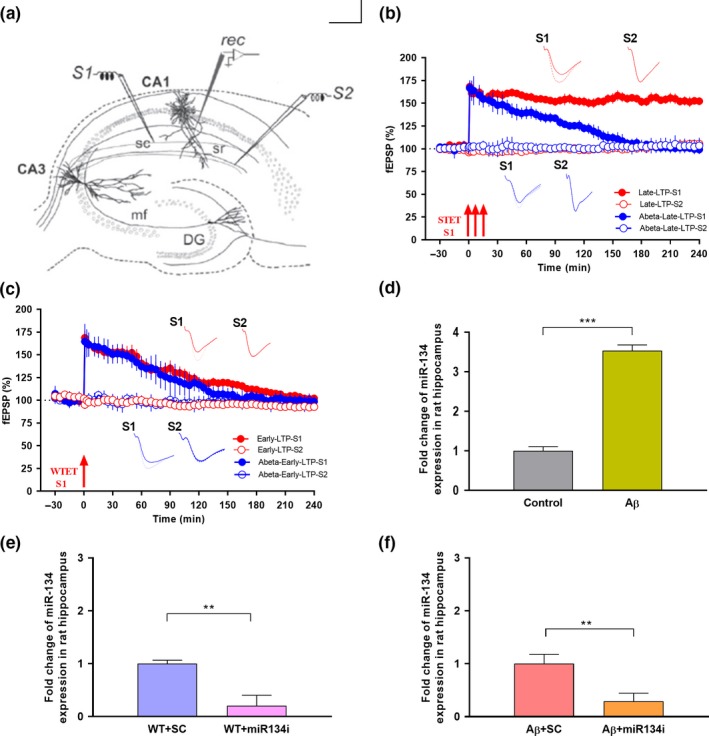
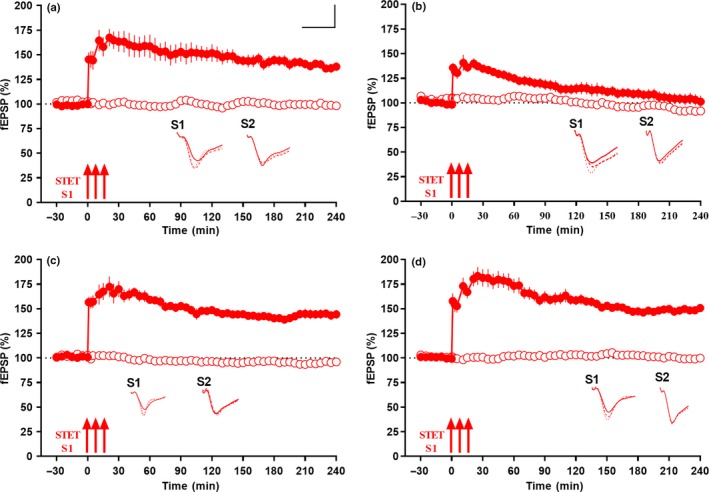
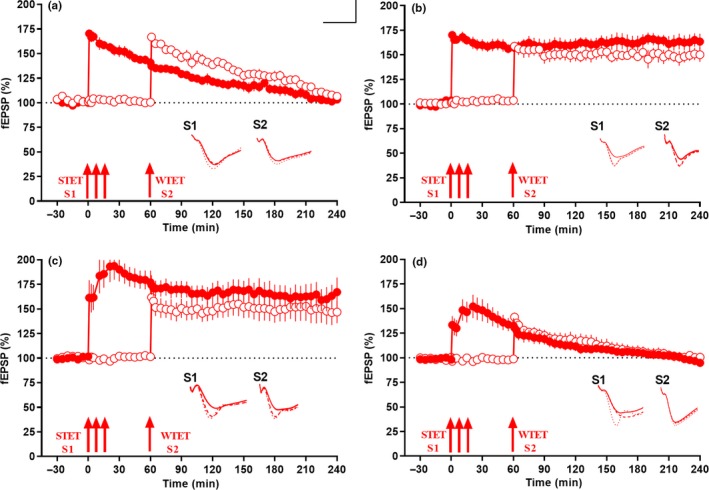
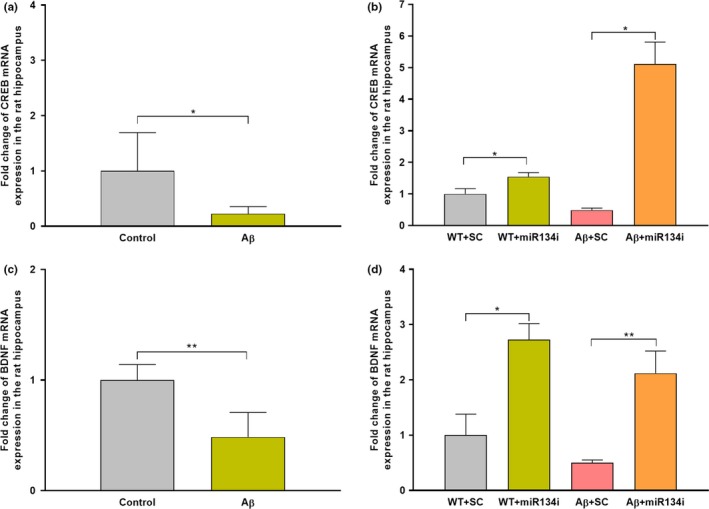
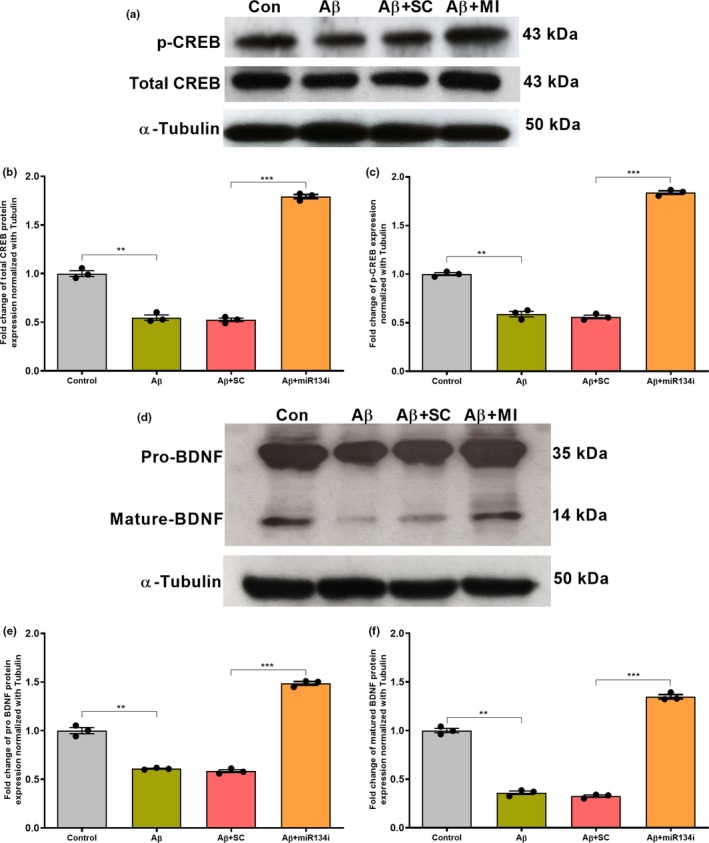
Progressive memory loss is one of the most common characteristics of Alzheimer's disease (AD), which has been shown to be caused by several factors including accumulation of amyloid β peptide (Aβ) plaques and neurofibrillary tangles. Synaptic plasticity and associative plasticity, the cellular basis of memory, are impaired in AD. Recent studies suggest a functional relevance of microRNAs (miRNAs) in regulating plasticity changes in AD, as their differential expressions were reported in many AD brain regions. However, the specific role of these miRNAs in AD has not been elucidated. We have reported earlier that late long-term potentiation (late LTP) and its associative mechanisms such as synaptic tagging and capture (STC) were impaired in Aβ (1-42)-induced AD condition. This study demonstrates that expression of miR-134-5p, a brain-specific miRNA is upregulated in Aβ (1-42)-treated AD hippocampus. Interestingly, the loss of function of miR-134-5p restored late LTP and STC in AD. In AD brains, inhibition of miR-134-5p elevated the expression of plasticity-related proteins (PRPs), cAMP-response-element binding protein (CREB-1) and brain-derived neurotrophic factor (BDNF), which are otherwise downregulated in AD condition. The results provide the first evidence that the miR-134-mediated post-transcriptional regulation of CREB-1 and BDNF is an important molecular mechanism underlying the plasticity deficit in AD; thus demonstrating the critical role of miR-134-5p as a potential therapeutic target for restoring plasticity in AD condition.
Keywords: Alzheimer's disease; Aβ(1-42); brain-derived neurotrophic factor; cAMP response element-binding protein; long-term potentiation; miRNA; synaptic tagging.
© 2019 The Authors. Aging Cell published by the Anatomical Society and John Wiley & Sons Ltd.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Alzheimer’s Association , (2015). 2015 Alzheimer's disease facts and figures. Alzheimer's & Dementia: The Journal of the Alzheimer's Association, 11(3), 332. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
