

Functional characterization of a novel PBX1 de novo missense variant identified in a patient with syndromic congenital heart disease
- PMID: 31625560
- PMCID: PMC7206850
- DOI: 10.1093/hmg/ddz231
Functional characterization of a novel PBX1 de novo missense variant identified in a patient with syndromic congenital heart disease
Abstract
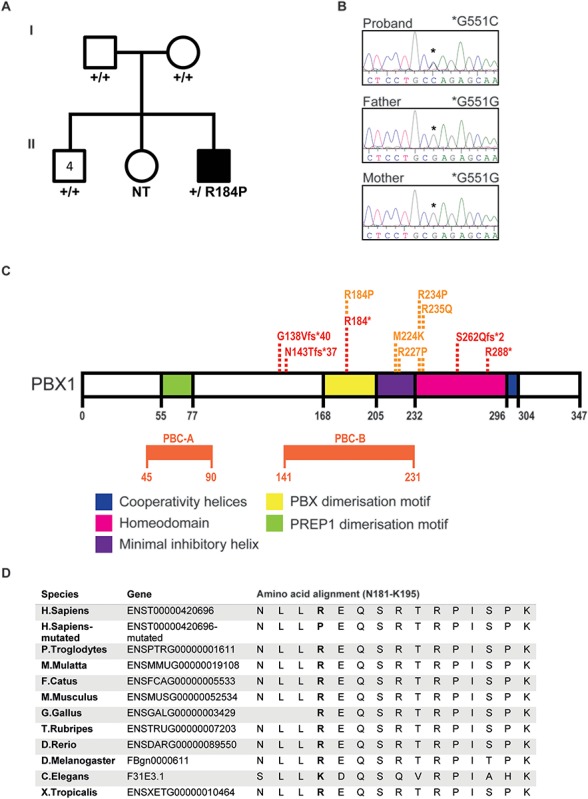
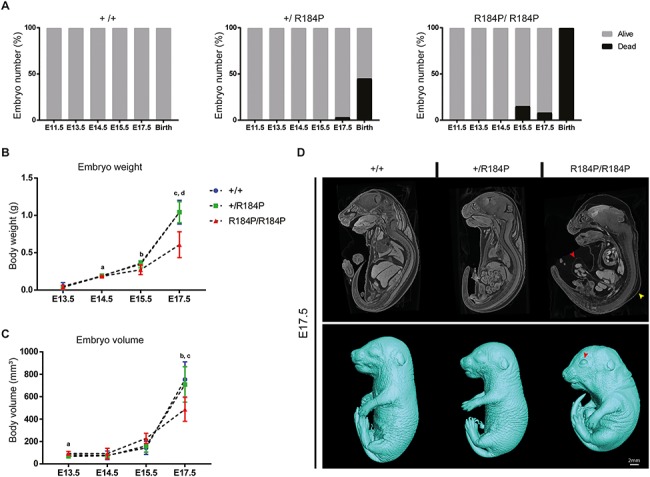
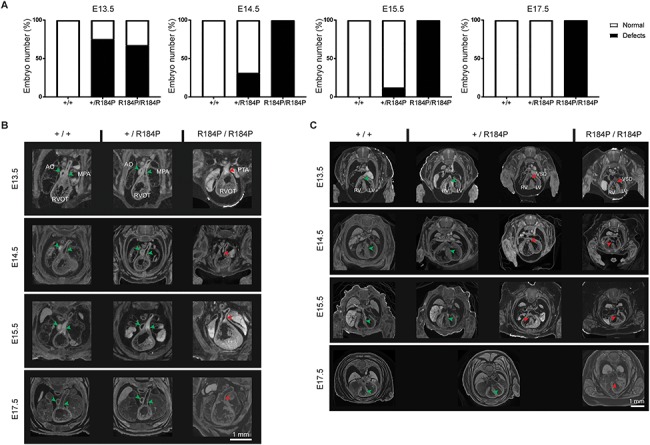
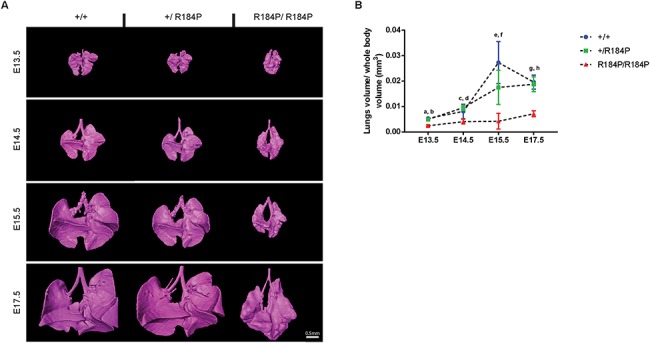
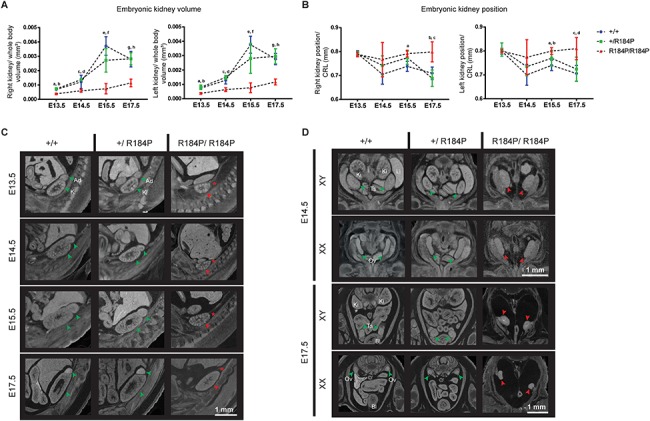
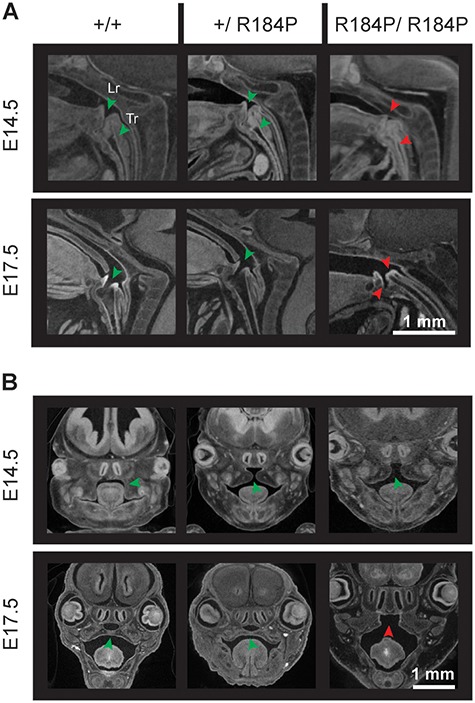
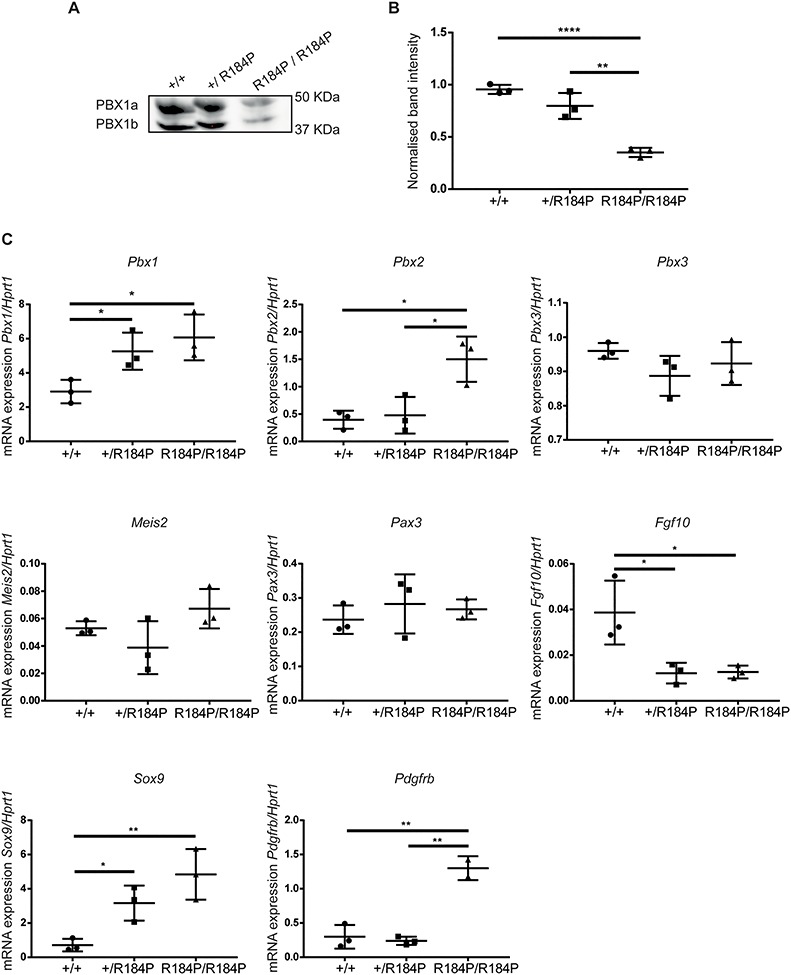
Pre-B cell leukemia factor 1 (PBX1) is an essential developmental transcription factor, mutations in which have recently been associated with CAKUTHED syndrome, characterized by multiple congenital defects including congenital heart disease (CHD). During analysis of a whole-exome-sequenced cohort of heterogeneous CHD patients, we identified a de novo missense variant, PBX1:c.551G>C p.R184P, in a patient with tetralogy of Fallot with absent pulmonary valve and extra-cardiac phenotypes. Functional analysis of this variant by creating a CRISPR-Cas9 gene-edited mouse model revealed multiple congenital anomalies. Congenital heart defects (persistent truncus arteriosus and ventricular septal defect), hypoplastic lungs, hypoplastic/ectopic kidneys, aplastic adrenal glands and spleen, as well as atretic trachea and palate defects were observed in the homozygous mutant embryos at multiple stages of development. We also observed developmental anomalies in a proportion of heterozygous embryos, suggestive of a dominant mode of inheritance. Analysis of gene expression and protein levels revealed that although Pbx1 transcripts are higher in homozygotes, amounts of PBX1 protein are significantly decreased. Here, we have presented the first functional model of a missense PBX1 variant and provided strong evidence that p.R184P is disease-causal. Our findings also expand the phenotypic spectrum associated with pathogenic PBX1 variants in both humans and mice.
© The Author(s) 2019. Published by Oxford University Press. All rights reserved. For permissions, please e-mail: journals.permission@oup.com.
Figures
References
-
- van der Linde D., Konings E.E.M., Slager M.A., Witsenburg M., Helbing W.A., Takkenberg J.J.M. and Roos-Hesselink J.W. (2011) Birth prevalence of congenital heart disease worldwide. J Am. Coll. Cardiol., 58, 2241–2247. - PubMed
-
- Blue G.M., Kirk E.P., Giannoulatou E., Sholler G.F., Dunwoodie S.L., Harvey R.P. and Winlaw D.S. (2017) Advances in the genetics of congenital heart disease a Clinician's guide. J. Am. Coll. Cardiol., 69, 860–870. - PubMed
-
- Alankarage D., Ip E., Szot J.O., Munro J., Blue G.M., Harrison K., Cuny H., Enriquez A., Troup M., Humphreys D.T. et al. (2019) Identification of clinically actionable variants from genome sequencing of families with congenital heart disease. Genetics In Medicine., 21, 1111–1120. - PubMed
-
- Szot J.O., Cuny H., Blue G.M., Humphreys D.T., Ip E., Harrison K., Sholler G.F., Giannoulatou E., Leo P., Duncan E.L. et al. (2018) A screening approach to identify clinically actionable variants causing congenital heart disease in exome data. Circ. Genom. Precis. Med., 11:e001978. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
