

Model system identification of novel congenital heart disease gene candidates: focus on RPL13
- PMID: 31625562
- PMCID: PMC7202142
- DOI: 10.1093/hmg/ddz213
Model system identification of novel congenital heart disease gene candidates: focus on RPL13
Abstract
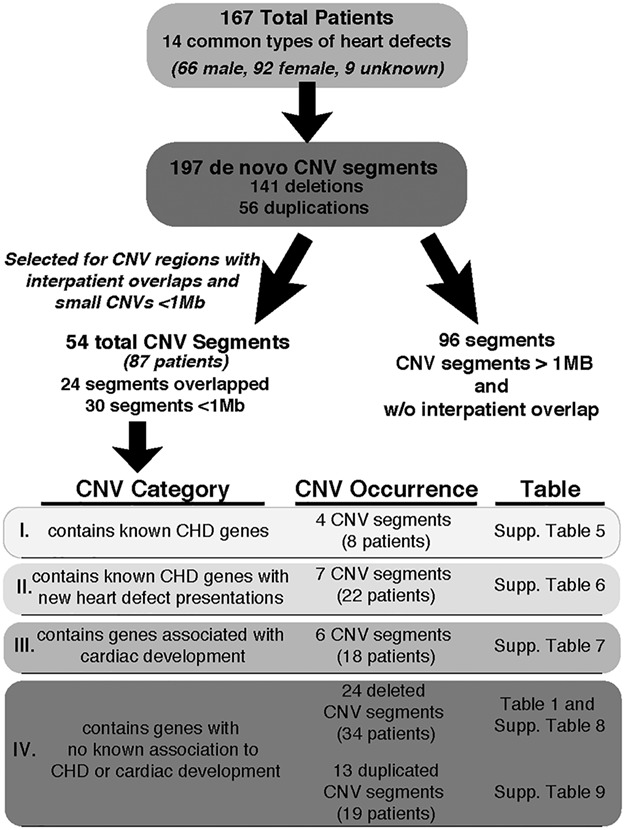
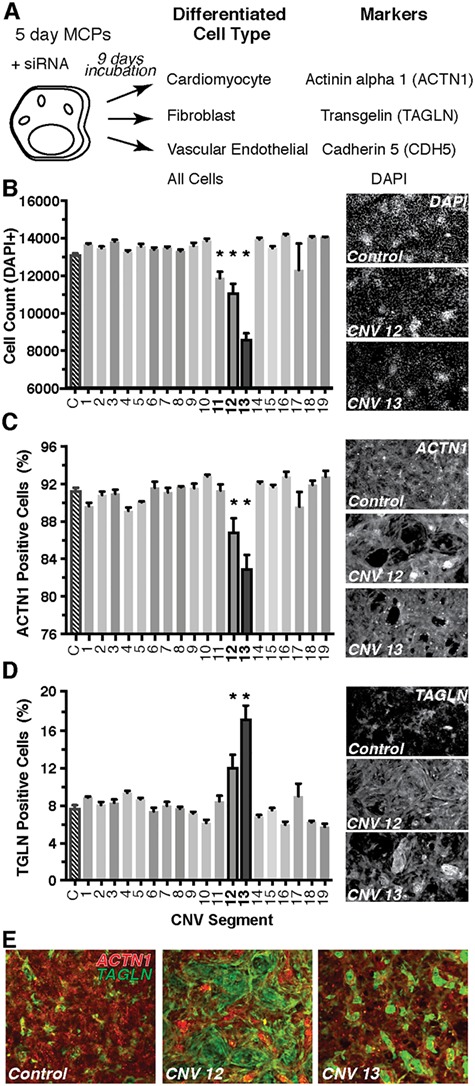
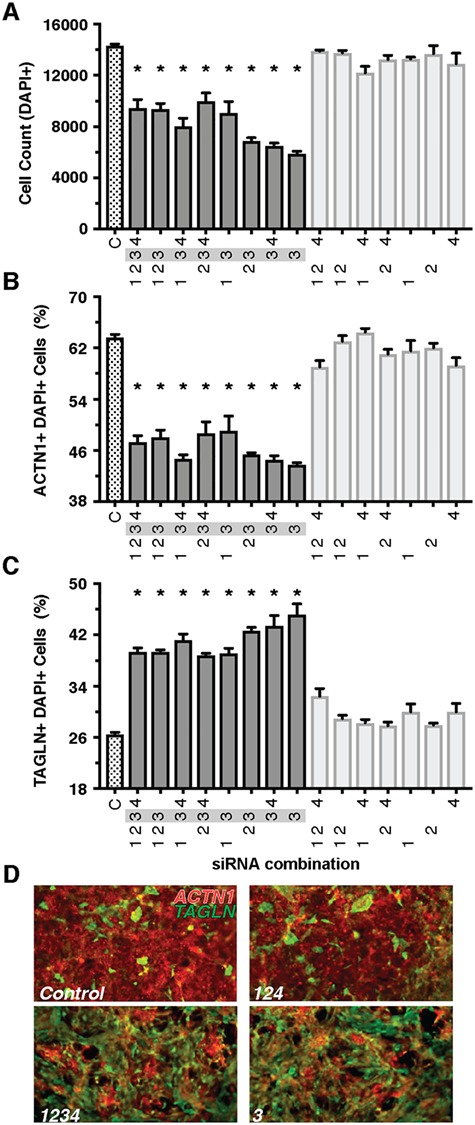
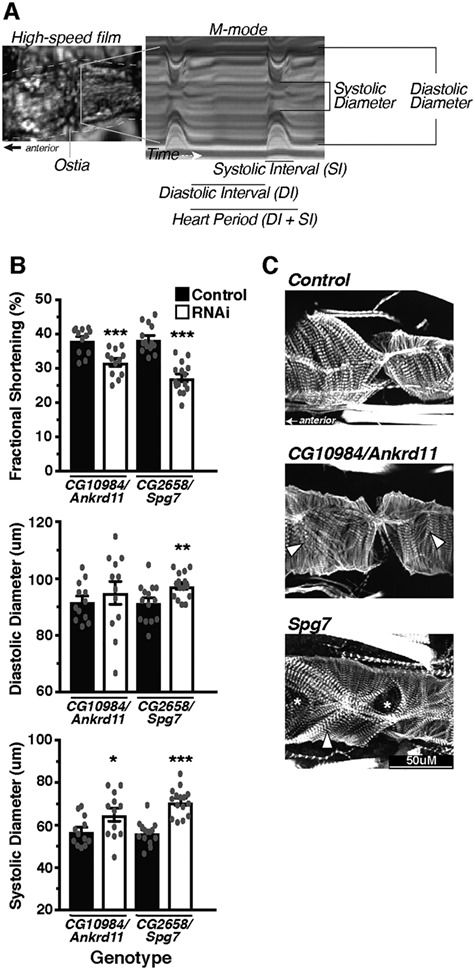
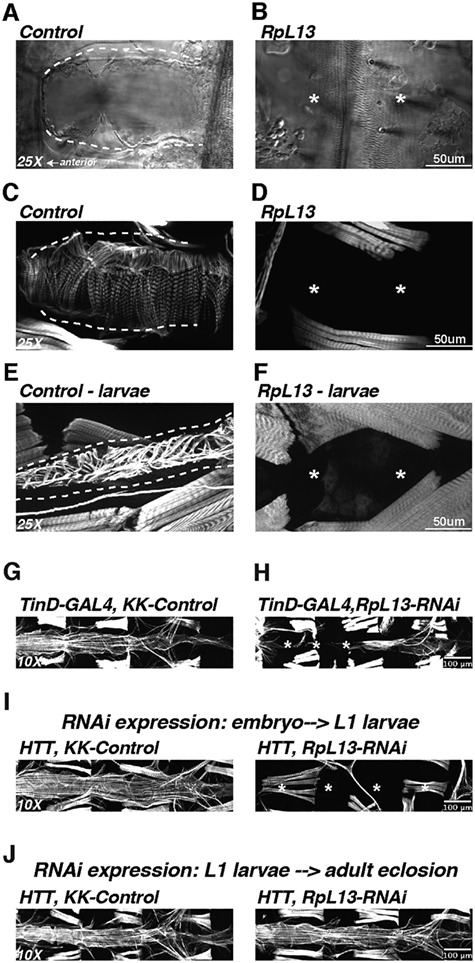
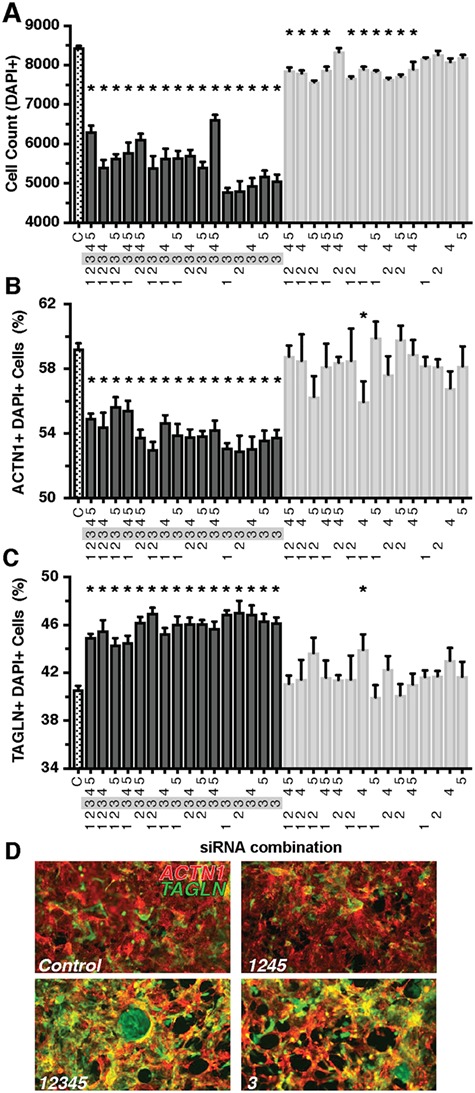
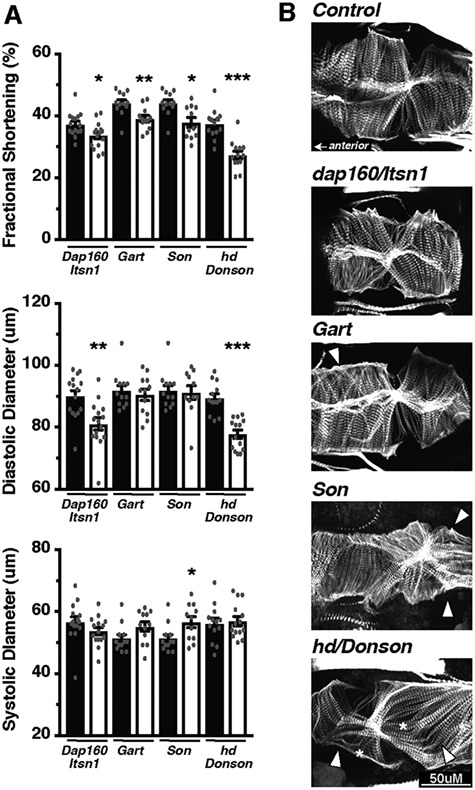
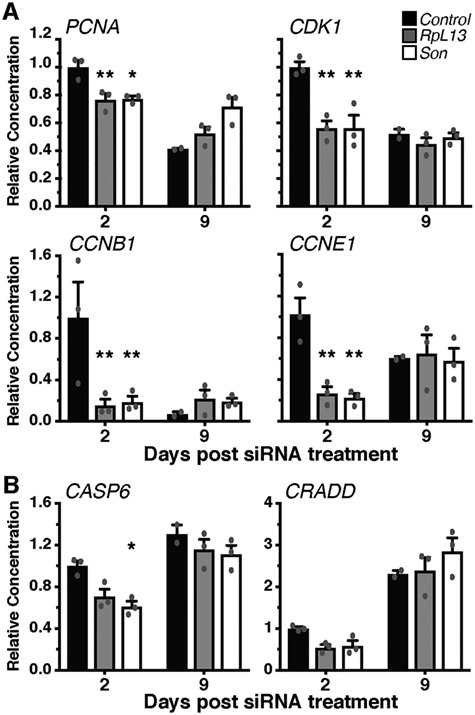
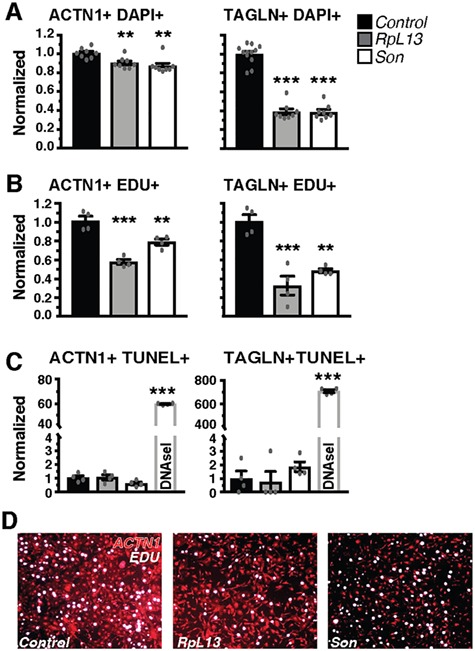
Genetics is a significant factor contributing to congenital heart disease (CHD), but our understanding of the genetic players and networks involved in CHD pathogenesis is limited. Here, we searched for de novo copy number variations (CNVs) in a cohort of 167 CHD patients to identify DNA segments containing potential pathogenic genes. Our search focused on new candidate disease genes within 19 deleted de novo CNVs, which did not cover known CHD genes. For this study, we developed an integrated high-throughput phenotypical platform to probe for defects in cardiogenesis and cardiac output in human induced pluripotent stem cell (iPSC)-derived multipotent cardiac progenitor (MCPs) cells and, in parallel, in the Drosophila in vivo heart model. Notably, knockdown (KD) in MCPs of RPL13, a ribosomal gene and SON, an RNA splicing cofactor, reduced proliferation and differentiation of cardiomyocytes, while increasing fibroblasts. In the fly, heart-specific RpL13 KD, predominantly at embryonic stages, resulted in a striking 'no heart' phenotype. KD of Son and Pdss2, among others, caused structural and functional defects, including reduced or abolished contractility, respectively. In summary, using a combination of human genetics and cardiac model systems, we identified new genes as candidates for causing human CHD, with particular emphasis on ribosomal genes, such as RPL13. This powerful, novel approach of combining cardiac phenotyping in human MCPs and in the in vivo Drosophila heart at high throughput will allow for testing large numbers of CHD candidates, based on patient genomic data, and for building upon existing genetic networks involved in heart development and disease.
© The Author(s) 2019. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
References
-
- Dolk H., Loane M., Garne E. and European Surveillance of Congenital Anomalies Working, G (2011) Congenital heart defects in Europe: prevalence and perinatal mortality, 2000 to 2005. Circulation, 123, 841–849. - PubMed
-
- Khoshnood B., de Vigan C., Vodovar V., Goujard J., Lhomme A., Bonnet D. and Goffinet F. (2006) Trends in antenatal diagnosis, pregnancy termination and perinatal mortality in infants with congenital heart disease: evaluation in the general population of Paris 1983-2000. J. Gynecol. Obstet. Biol. Reprod. (Paris), 35, 455–464. - PubMed
-
- Khairy P., Ionescu-Ittu R., Mackie A.S., Abrahamowicz M., Pilote L. and Marelli A.J. (2010) Changing mortality in congenital heart disease. J. Am. Coll. Cardiol., 56, 1149–1157. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
