

A missense mutation in the catalytic domain of O-GlcNAc transferase links perturbations in protein O-GlcNAcylation to X-linked intellectual disability
- PMID: 31627256
- PMCID: PMC7042088
- DOI: 10.1002/1873-3468.13640
A missense mutation in the catalytic domain of O-GlcNAc transferase links perturbations in protein O-GlcNAcylation to X-linked intellectual disability
Abstract
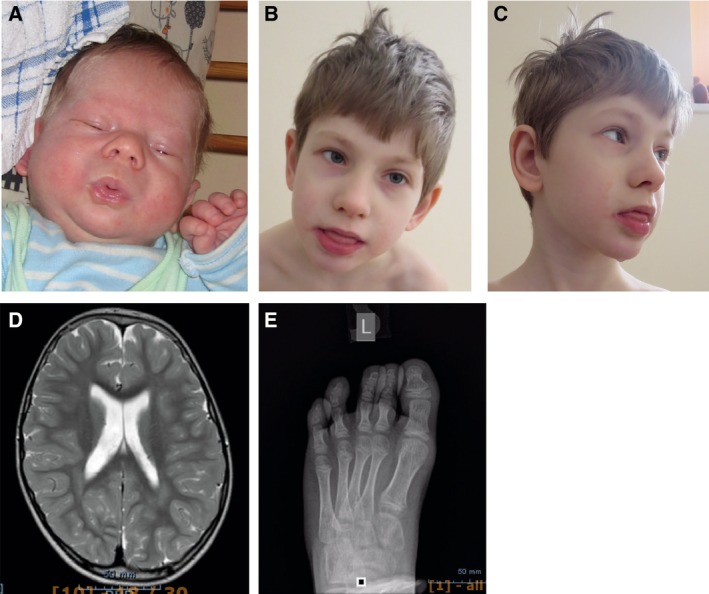
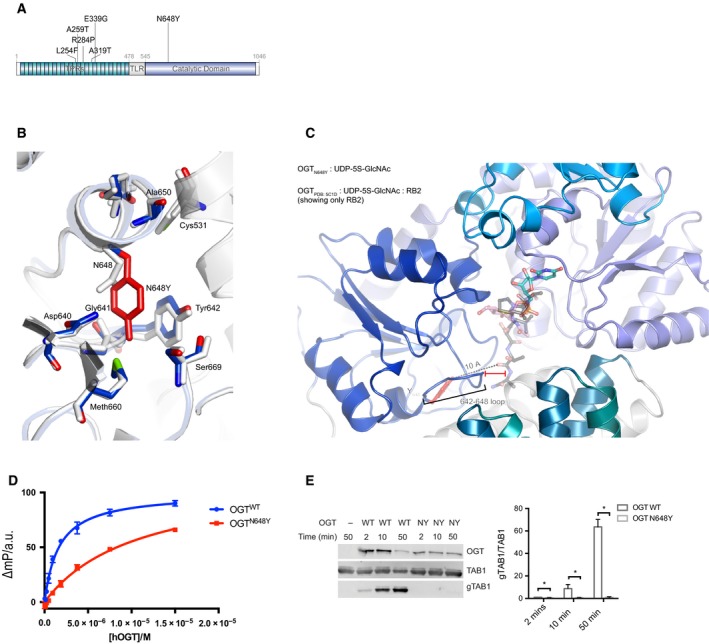
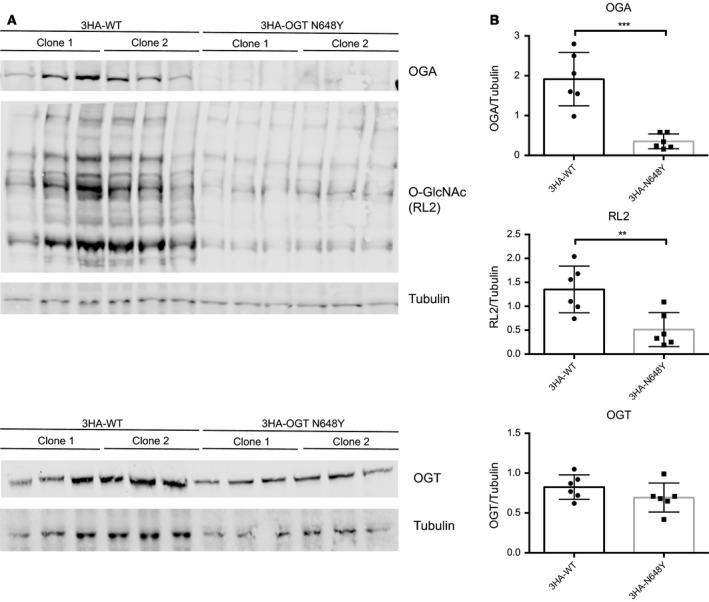
X-linked intellectual disabilities (XLID) are common developmental disorders. The enzyme O-GlcNAc transferase encoded by OGT, a recently discovered XLID gene, attaches O-GlcNAc to nuclear and cytoplasmic proteins. As few missense mutations have been described, it is unclear what the aetiology of the patient phenotypes is. Here, we report the discovery of a missense mutation in the catalytic domain of OGT in an XLID patient. X-ray crystallography reveals that this variant leads to structural rearrangements in the catalytic domain. The mutation reduces in vitro OGT activity on substrate peptides/protein. Mouse embryonic stem cells carrying the mutation reveal reduced O-GlcNAcase (OGA) and global O-GlcNAc levels. These data suggest a direct link between changes in the O-GlcNAcome and intellectual disability observed in patients carrying OGT mutations.
Keywords: O-GlcNAc; OGT; OGlcNAC transferase; XLID; intellectual disability; neurodevelopment.
© 2019 The Authors. FEBS Letters published by John Wiley & Sons Ltd on behalf of Federation of European Biochemical Societies.
Figures
References
-
- Maulik PK, Mascarenhas MN, Mathers CD, Dua T and Saxena S (2011) Prevalence of intellectual disability: a meta‐analysis of population‐based studies. Res Dev Disabil 32, 419–436. - PubMed
-
- American Psychiatric Association (2013) Diagnostic and Statistical Manual of Mental Disorders. American Psychiatric Association, Washington, D.C.
-
- Kvarnung M and Nordgren A (2017) Intellectual disability & rare disorders: a diagnostic challenge. Adv Exp Med Biol 1031, 39–54. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
