

DeepImpute: an accurate, fast, and scalable deep neural network method to impute single-cell RNA-seq data
- PMID: 31627739
- PMCID: PMC6798445
- DOI: 10.1186/s13059-019-1837-6
DeepImpute: an accurate, fast, and scalable deep neural network method to impute single-cell RNA-seq data
Abstract
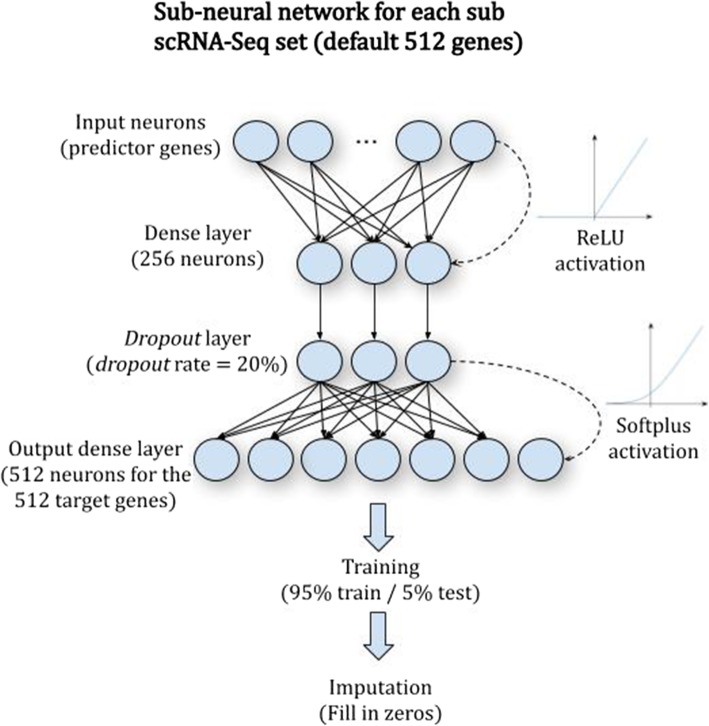
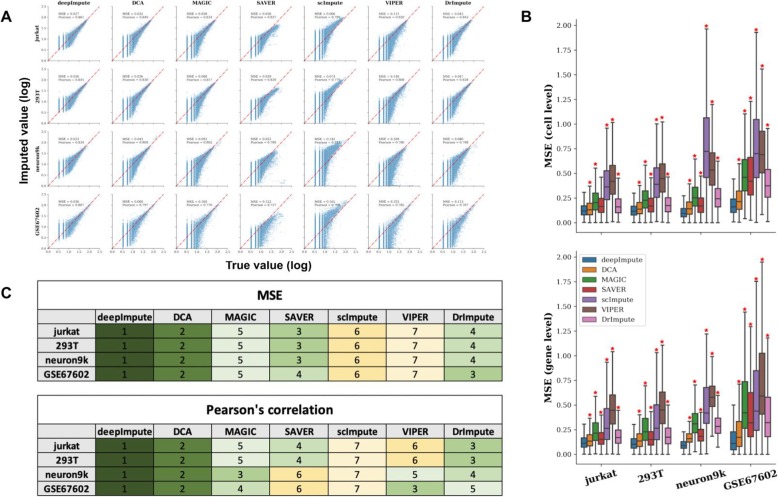
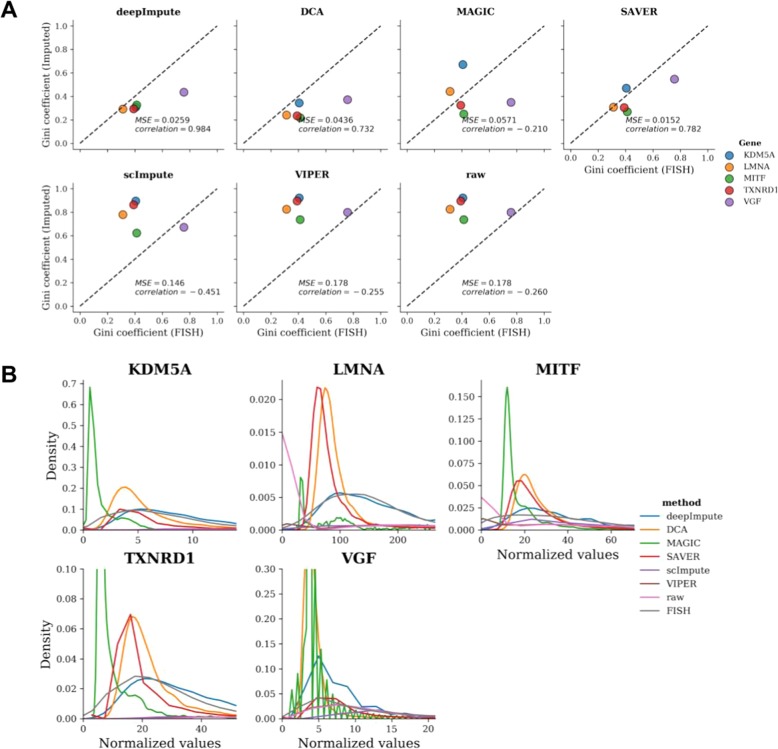
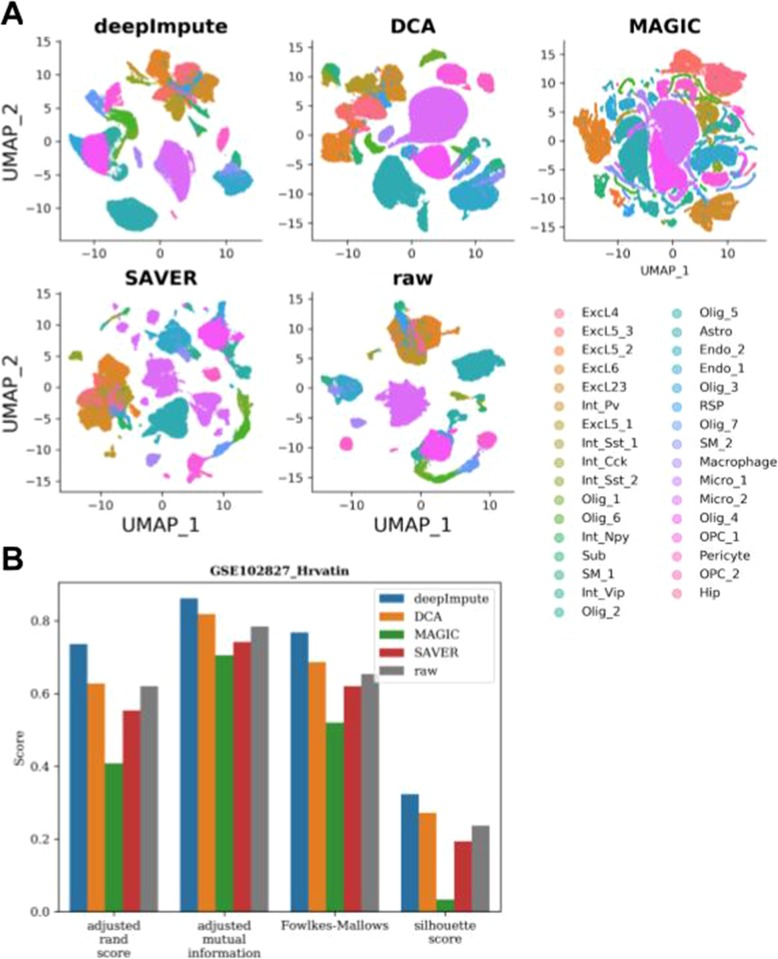
Single-cell RNA sequencing (scRNA-seq) offers new opportunities to study gene expression of tens of thousands of single cells simultaneously. We present DeepImpute, a deep neural network-based imputation algorithm that uses dropout layers and loss functions to learn patterns in the data, allowing for accurate imputation. Overall, DeepImpute yields better accuracy than other six publicly available scRNA-seq imputation methods on experimental data, as measured by the mean squared error or Pearson's correlation coefficient. DeepImpute is an accurate, fast, and scalable imputation tool that is suited to handle the ever-increasing volume of scRNA-seq data, and is freely available at https://github.com/lanagarmire/DeepImpute .
Keywords: Deep learning; DeepImpute; Dropout; Imputation; Machine learning; Neural network; RNA-seq; Single-cell.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
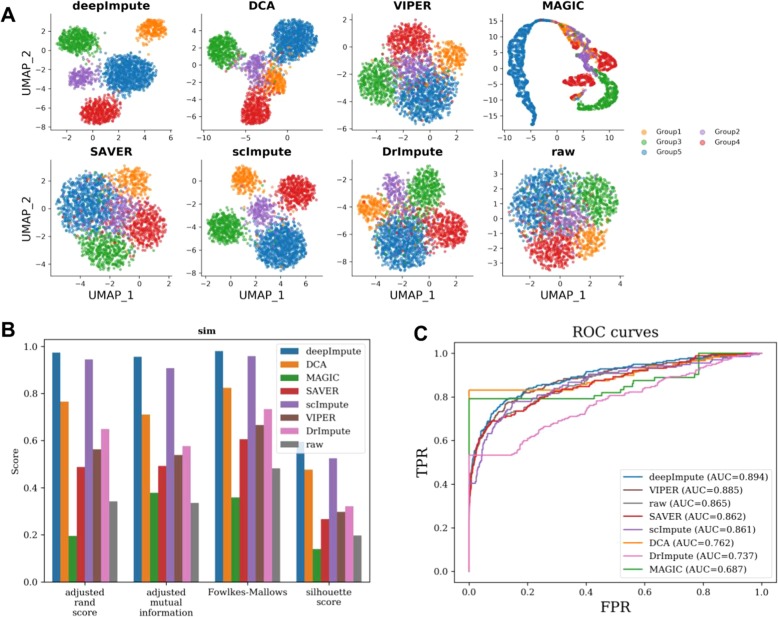
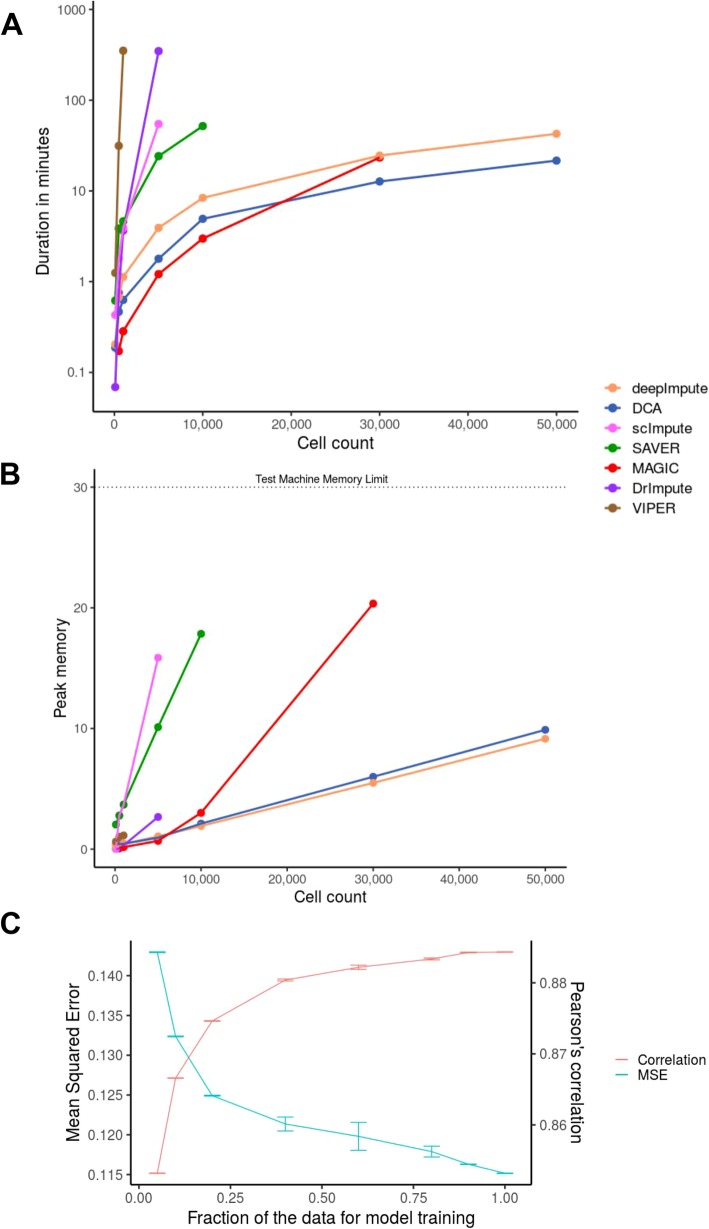
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
