

Creatine uptake regulates CD8 T cell antitumor immunity
- PMID: 31628186
- PMCID: PMC6888972
- DOI: 10.1084/jem.20182044
Creatine uptake regulates CD8 T cell antitumor immunity
Abstract
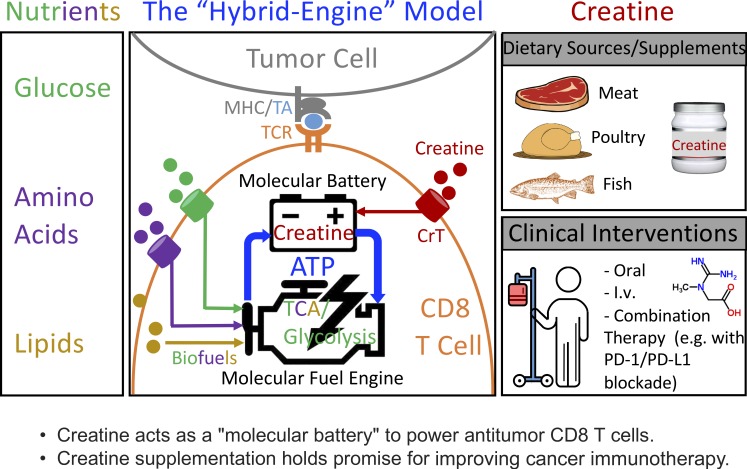
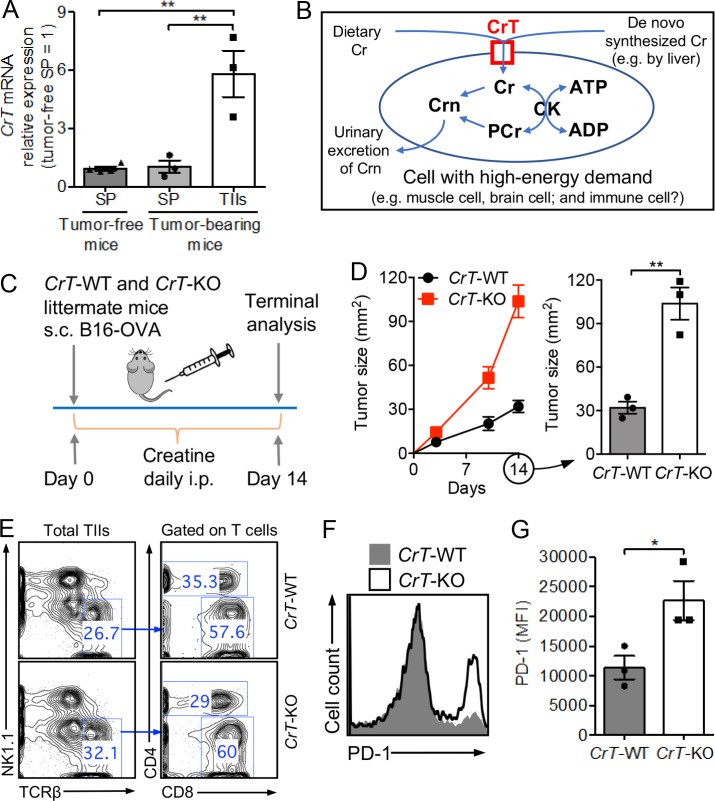
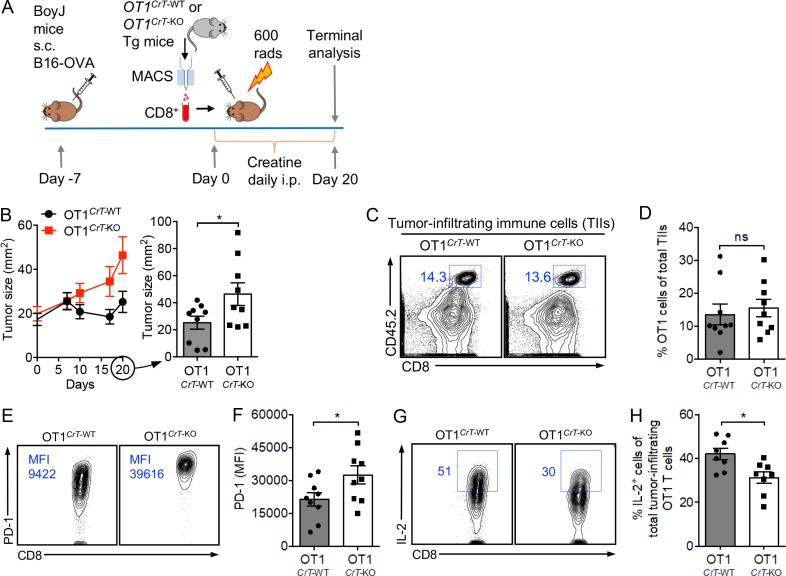
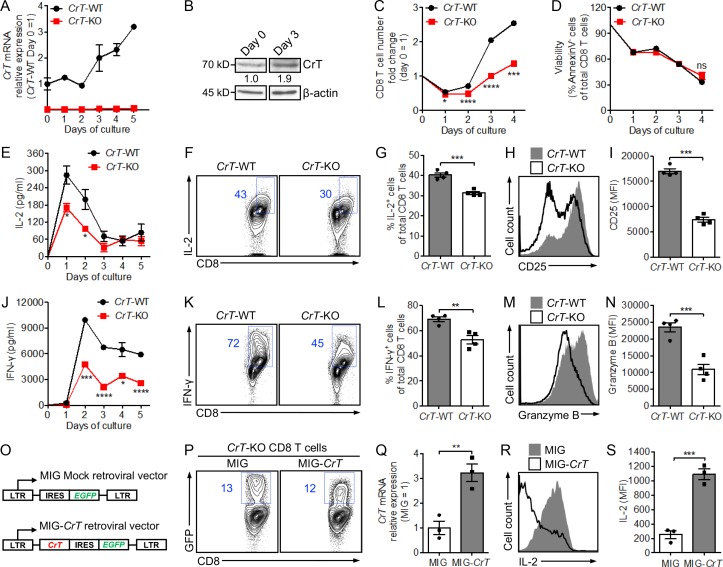
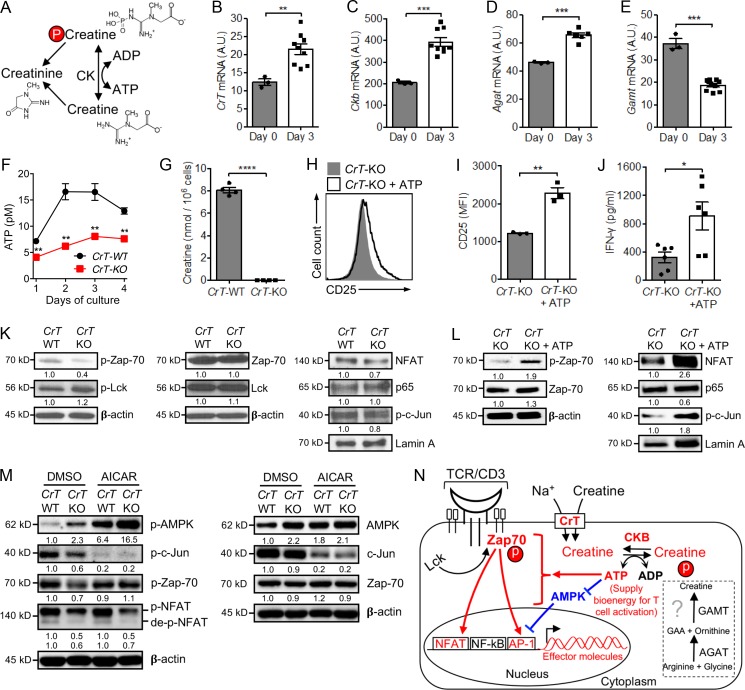
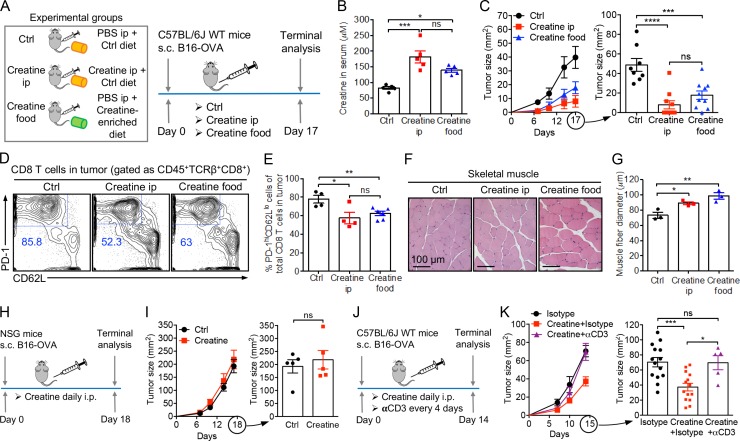
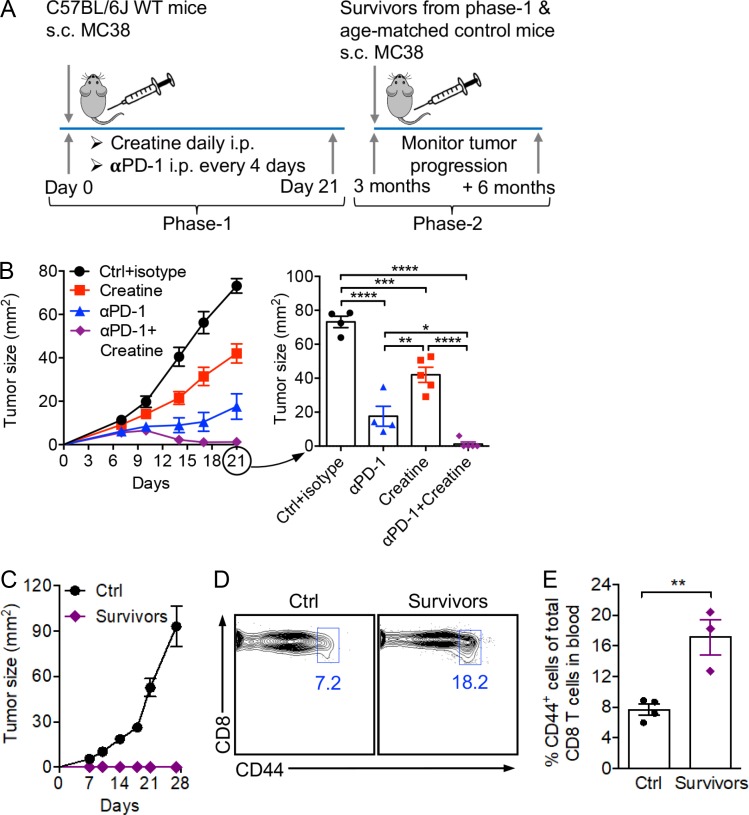
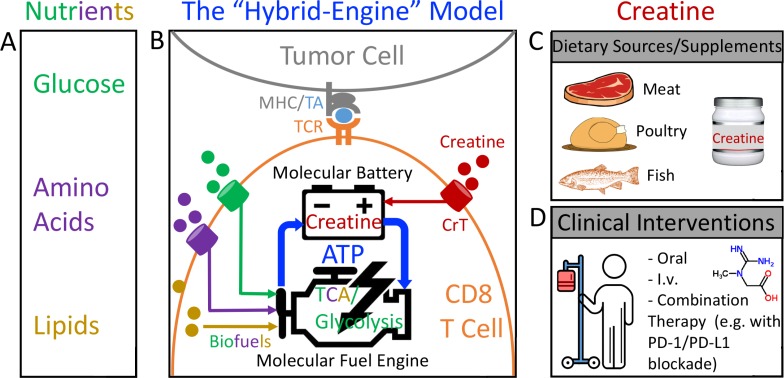
T cells demand massive energy to combat cancer; however, the metabolic regulators controlling antitumor T cell immunity have just begun to be unveiled. When studying nutrient usage of tumor-infiltrating immune cells in mice, we detected a sharp increase of the expression of a CrT (Slc6a8) gene, which encodes a surface transporter controlling the uptake of creatine into a cell. Using CrT knockout mice, we showed that creatine uptake deficiency severely impaired antitumor T cell immunity. Supplementing creatine to WT mice significantly suppressed tumor growth in multiple mouse tumor models, and the combination of creatine supplementation with a PD-1/PD-L1 blockade treatment showed synergistic tumor suppression efficacy. We further demonstrated that creatine acts as a "molecular battery" conserving bioenergy to power T cell activities. Therefore, our results have identified creatine as an important metabolic regulator controlling antitumor T cell immunity, underscoring the potential of creatine supplementation to improve T cell-based cancer immunotherapies.
© 2019 Di Biase et al.
Figures
References
-
- Bengsch B., Johnson A.L., Kurachi M., Odorizzi P.M., Pauken K.E., Attanasio J., Stelekati E., McLane L.M., Paley M.A., Delgoffe G.M., and Wherry E.J.. 2016. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8(+) T Cell Exhaustion. Immunity. 45:358–373. 10.1016/j.immuni.2016.07.008 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
