

Oxidative DNA damage is concurrently repaired by base excision repair (BER) and apyrimidinic endonuclease 1 (APE1)-initiated nonhomologous end joining (NHEJ) in cortical neurons
- PMID: 31628877
- PMCID: PMC7317839
- DOI: 10.1111/nan.12584
Oxidative DNA damage is concurrently repaired by base excision repair (BER) and apyrimidinic endonuclease 1 (APE1)-initiated nonhomologous end joining (NHEJ) in cortical neurons
Abstract
Aims: Accumulating studies have suggested that base excision repair (BER) is the major repair pathway of oxidative DNA damage in neurons, and neurons are deficient in other DNA repair pathways, including nucleotide excision repair and homologous recombination repair. However, some studies have demonstrated that neurons could efficiently repair glutamate- and menadione-induced double-strand breaks (DSBs), suggesting that the DSB repair mechanisms might be implicated in neuronal health. In this study, we hypothesized that BER and nonhomologous end joining (NHEJ) work together to repair oxidative DNA damage in neurons.
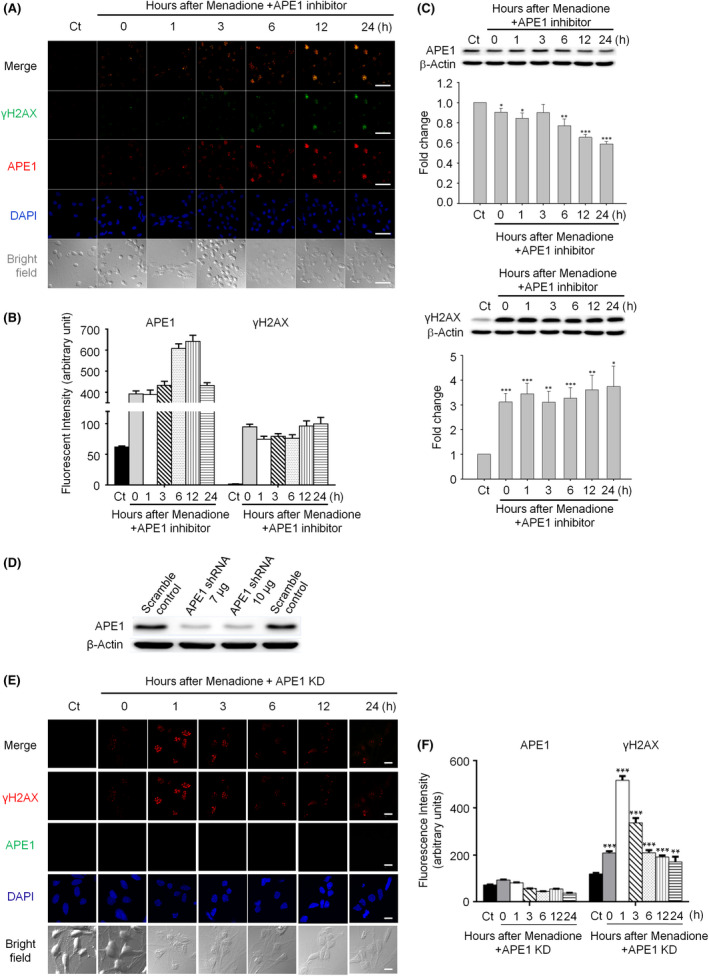
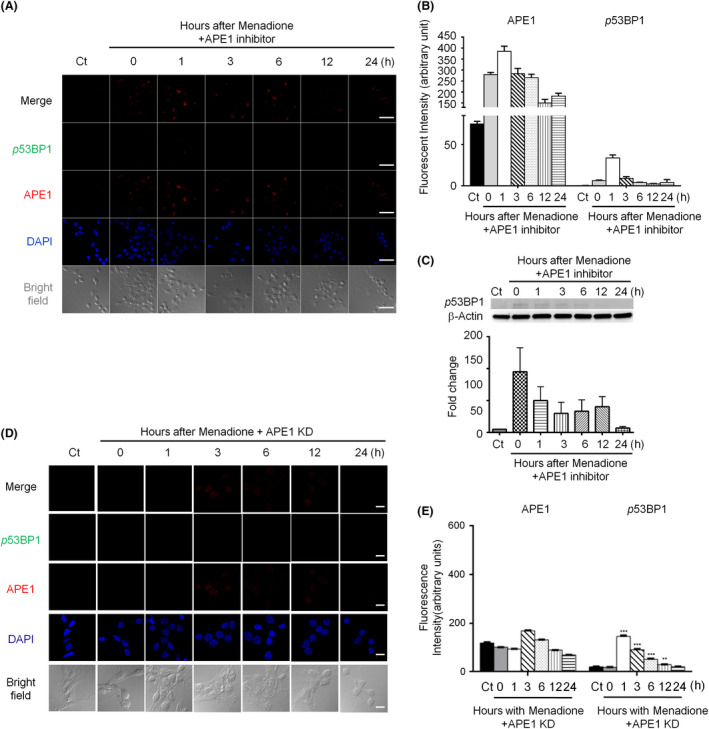
Methods: Immunohistochemistry and confocal microscopy were employed to examine the colocalization of apyrimidinic endonuclease 1 (APE1), histone variant 2AX (γH2AX) and phosphorylated p53-binding protein (53BP1). APE1 inhibitor and shRNA were respectively applied to suppress APE1 activity and protein expression to determine the correlation of APE1 and DSB formation. The neutral comet assay was used to determine and quantitate the formation of DSB.
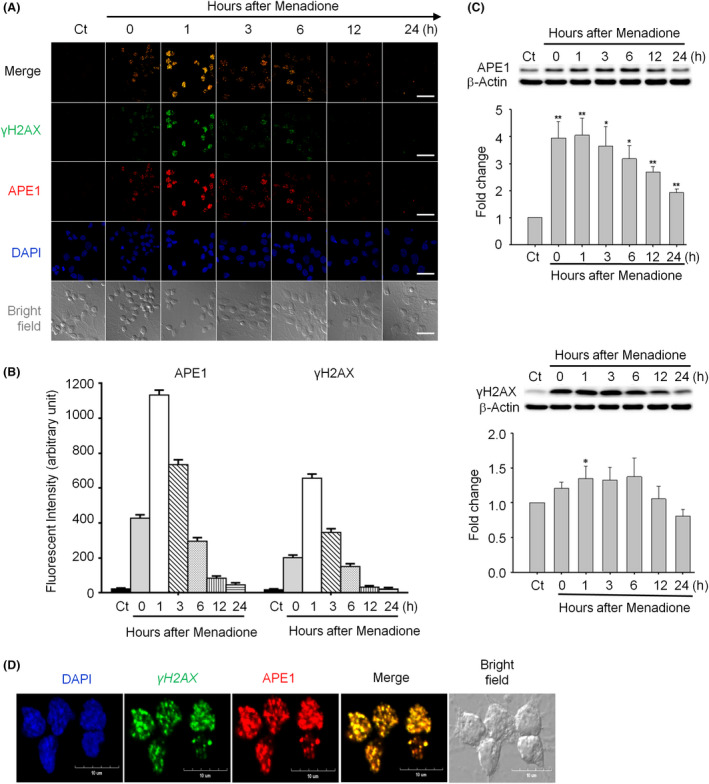
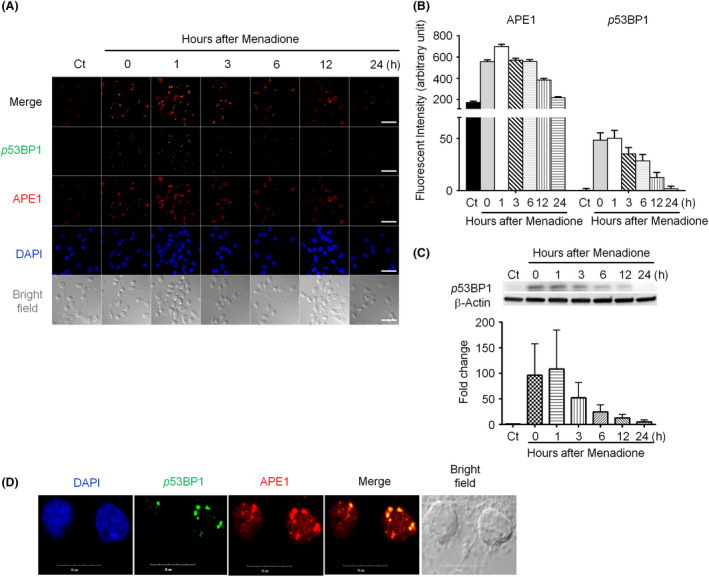
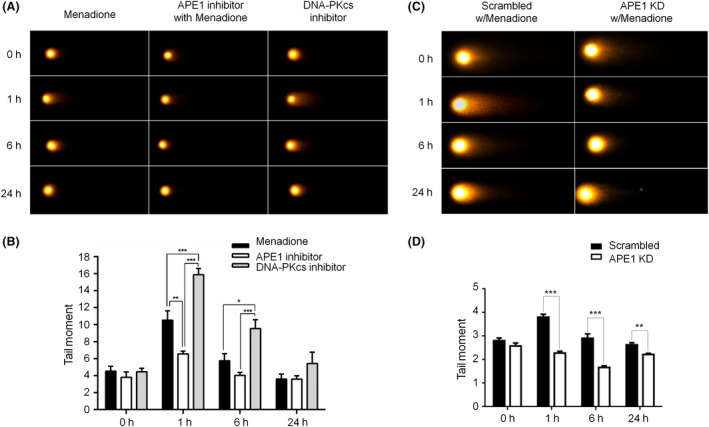
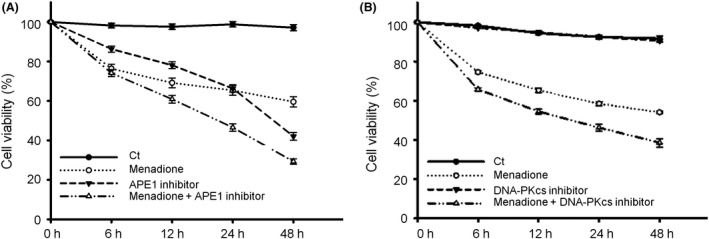
Results: Both γH2AX and 53BP1 were upregulated and colocalized with APE1 in the nuclei of rat cortical neurons subjected to menadione-induced oxidative insults. Phospho53BP1 foci were efficiently abolished, but γH2AX foci persisted following the suppression of APE1 activity. Comet assays demonstrated that the inhibition of APE1 decreased the DSB formation.
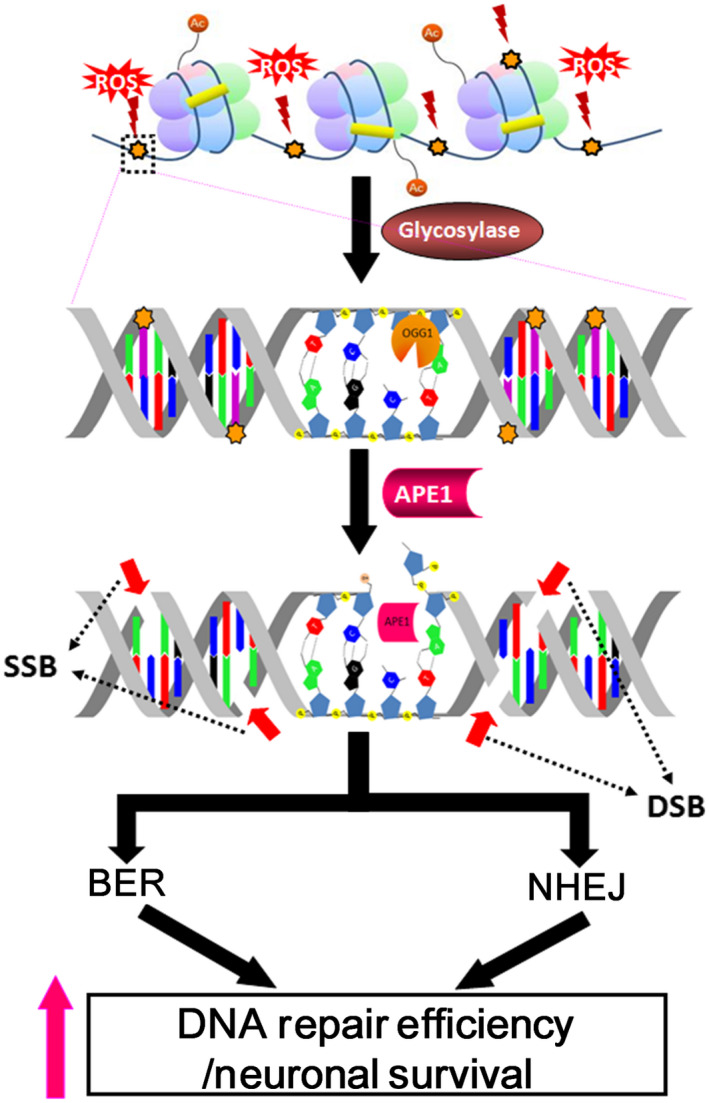
Conclusions: Our results indicate that APE1 can engage the NHEJ mechanism in the repair of oxidative DNA damage in neurons. These findings provide insights into the mechanisms underlying the efficient repair of oxidative DNA damage in neurons despite the high oxidative burden.
Keywords: apurinic/apyrimidinic endonuclease 1 (APE1); base excision repair (BER); neuron; nonhomologous end joining (NHEJ); oxidative DNA damage.
© 2019 The Authors. Neuropathology and Applied Neurobiology published by John Wiley & Sons Ltd on behalf of British Neuropathological Society.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Bohr VA, Ottersen OP, Tonjum T. Genome instability and DNA repair in brain, ageing and neurological disease. Neuroscience 2007; 145: 1183–6 - PubMed
-
- Mandavilli BS, Rao KS. Accumulation of DNA damage in aging neurons occurs through a mechanism other than apoptosis. J. Neurochem 1996; 67: 1559–65 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
